Abstract
Methylphenidate (MPD) is psychostimulant, similar to cocaine and amphetamine, that is commonly used to treat attention deficit hyperactivity disorder and is increasingly being abused by healthy subjects for its psychoactive effects such as memory retention cognitive enhancement for young, adult and the elderly and recreation. MPD’s action on the brain reward/motive circuit is still under investigation, however it is known that in animals chronic use of MPD leads to behavioral sensitization, an experimental indicator associated with dependence. To investigate this neural circuit’s role in response to acute and chronic MPD, three different lesions (non-specific, dopaminergic specific, and glutaminergic specific lesions) have been conducted at the nucleus accumbens (NAc), the ventral tegmental area (VTA), the caudate nucleus (CN), and the prefrontal cortex (PFC), to assess the structure, dopaminergic signaling, and glutaminergic signaling roles in response to MPD. The three types of lesions show that each one of the above four brain areas participate differently in the acute and chronic effect of MPD and have helped determine which type of signaling is critical for the acute and/or chronic behavioral adaptions to MPD.
Keywords
Methylphenidate, Specific lesions, VTA, NAc, CN, PFC, Motive circuit
Introduction
Methylphenidate (MPD; Ritalin®, Concerta®) is one of the first line treatments for patients with Attention Deficit Hyperactivity Disorder (ADHD), and is growing in illicit use for recreation or cognitive enhancement in normal subjects [1-6]. MPD has been shown to produce a variety of effects on the locomotive activity of normal and ADHD-model animals [7-23]. Acute exposure to MPD causes an increase in locomotor activity, while chronic use of MPD has been reported to produce behavioral tolerance [17,24,25], withdrawal [11], or sensitization [10, 17,19,21,25-27].
MPD shares a similar pharmacologic profile to other psychostimulants including amphetamine and cocaine [24,28-30]. It is effective in treating ADHD, in addition to disorders of alertness in children with learning difficulties and of sleep-wake cycles such as narcolepsy and chronic fatigue [31-33]. MPD has been shown to bind to the dopamine transporter preventing dopamine reuptake to the presynaptic terminal, similar to the mechanism of action of cocaine and methamphetamine [24,28,29]. Decreased dopamine reuptake increases dopamine in the synaptic cleft, leading to increased signaling in the postsynaptic neuron which underlies MPD’s status as an indirect dopamine agonist [29,34,35]. This increase in dopamine is believed to be linked to the reinforcing effect of psychostimulants such as MPD [36,37].
Acute administration of psychostimulants such as MPD in normal and ADHD model animals results in an increase in locomotor activity [23,25-27,38]. At low doses, this manifests as a net increase in locomotive activity while at high doses it becomes repetitive purples movements termed stereotypic behavior [39-43]. Chronic administration of psychostimulants results in neural plasticity that will ultimately lead to the dependent state. In animal models, tolerance, withdrawal, and behavioral sensitization are used as experimental bioindicators of the abusive potential of a compound [24-27,32,39,44-47]. Behavioral sensitization, or the progressive augmentation of the drug effect produced by re-administration of previous doses, has been implicated as the physiologic underpinning of the symptom of drug craving [48,49], and serves as one of the experimental biomarkers that the drug under investigation has properties consistent with a drug of abuse [24,26,27,45,47,50].
Underlying these maladaptive responses to the effects of MPD is the brain’s reward/motive circuit [49,51-54]. This circuit (Figure 1) is made up of central nervous system (CNS) structures that are members of the executive function, mesolimbic, and motor systems and work in concert to mediate the behavior of an organism [50,55] and includes the ventral tegmental area (VTA), the nucleus accumbens (NAc), the caudate nucleus (CN), and the pre-frontal cortex (PFC), [56,57]. Classically, the reward circuit is described as originating from the VTA which projected to the NAc, which then projected to the PFC [53,56,58]. However, there is now increased understanding that the projections from the VTA are diffuse to the ventral and dorsal striatum, including the CN, and that reciprocal connections between these and other CNS structures all contribute to the rewards system [53,56,58].
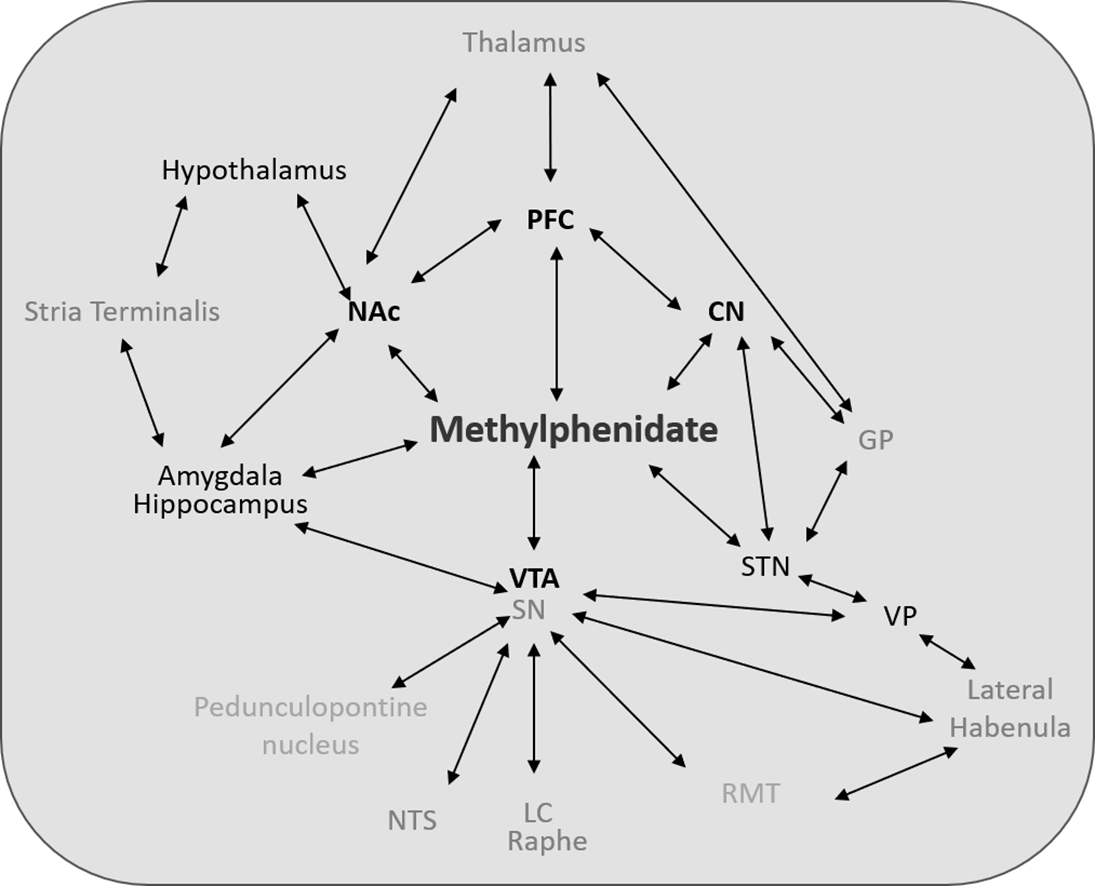
Figure 1. Figure 1 shows a representative diagram of the multiple interactions and pathways that mediate the acute and chronic effects of MPD in the rat CNS. Note that not all pathways are shown, nor is diagram meant to be anatomically to scale. Abbreviations not found in text: GP: Globus Pallidus; LC: Locus Ceruleus; NTS: Nucleus Tractus Solitarius; RMT: Rostromedial Tegmentum; STN: Subthalamic Nucleus; VP: Ventral Pallidum.
Understanding the neural pathways and their components that mediate the complex responses to MPD exposure remains incomplete. One of the methods used to study them is to ablate a specific brain area of an animal model’s CNS structure or signaling pathway (non-specifically or specifically) and challenge the subject to the substance. This manuscript reviews the non-specific electrolytic, dopaminergic specific, and glutaminergic specific lesions to the VTA (Figure 3) [59], to the NAc (Figure 4) [13,15,60], to the CN [4,7] (Figure 5) and to the PFC (Figure 6) [11,14,15], in the setting of acute and chronic MPD exposure that have been conducted. Bilateral non-specific lesions were conducted with electrolytic ablation [13,15,60], bilateral chemical dopaminergic specific lesions were obtained with local microinjection of the neurotoxin 6-hydroxydopamine (6-OHDA) [7,13,61-63], and bilateral glutaminergic specific ablation was obtained with local microinjection of the neurotoxin ibotenic acid [64-67] within the VTA, NAc, CN & PFC. Animal response to MPD was monitored and recorded with a computerized animal tracker in a behavioral open field assay, a means to track animal locomotive activity. It can track multiple movement parameters which can be analyzed by software to output distinct behavioral expression of locomotion which can then be followed over time to determine the acute and chronic effects of MPD. By drawing together current understandings of neural anatomy, circuitry, and these lesions’ effects, we hope to offer a perspective on the current knowledge governing the effects of MPD.
Methods
The methods used in this review were published in detail [4,7,11,13-15,59,60,68,69]. In short, 160 male Sprague-Dawley rats weighing 170-180 g were obtained from Harlan Labs (Indianapolis, IN, USA). Animals were individually placed in plexiglass cages (40.5 x 40.5 x 31.5 cm in dimension) in a soundproof room for 4-5 days to acclimate prior to experimentation. These cages served as the home and test cage (Figure 2). Animals were maintained on a 12-hour light/dark cycle that began at 06:00. Food and water were provided ad libitum throughout the experiment, and the temperature was kept at 21 ± 2°C with a relative humidity of 37-42%. Each of the four-brain lesion consist five group each N=8 as follow: 1). Intact animals; 2). Shame operated; 3). Nonspecific bilateral electrolytic lesion; 4). 6-OH DA bilateral injection for dopamine signaling ablation and 5). Ibotonic acid bilateral injection for chemical ablation of the glutaminergic system (Table 1). This protocol was approved by our Animal Welfare Committee and carried out in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals. On experimental day 1 (ED 1-Sal) animals were weighed and 0.8 mL of 0.9% saline was administered intra-peritoneal (i.p). All animals weighed 200-220 g at that time. Locomotive behavioral activity was recorded for 120 minutes post-injection to establish a baseline prior to surgical manipulation. On experimental day 2 (ED 2), the lesion and sham groups underwent surgery and were then allowed to recover for approximately 5 days (ED 3-7). On experimental day 8, saline was re-administered (ED 8-Sal) and post-surgical locomotor activity was recorded for 120 minutes to compare with the pre-surgical baseline (ED 1-Sal). Starting on experimental day 9 (ED 9-MPD), daily injections of 2.5 mg/kg MPD (Mallinckrodt, Hazelwood MO) dissolved in 0.8 mL of 0.9% saline were administer for 6 consecutive days (ED 9-MPD to ED 14-MPD), and activity recorded for 120 minutes post-injection. This dose of 2.5 mg/kg MPD has been shown to be sufficient to elicit behavioral sensitization in rats in previous dose-response experiments [7-9,17-23,70-72]. For the next 3 days (ED 15-17), animals received no injections (the washout period). After the washout period (ED 18-MPD), the rats were re-challenged with MPD at the previous dose of 2.5 mg/kg and behavioral activity was observed for 120 minutes (Table 1). On ED 2, the sham operation group, the electrolytic lesion group, the 6-OHDA group, and the ibotonic acid group animals were anaesthetized with 60 mg/ kg pentobarbital and placed in the stereotactic apparatus. An incision was to expose the skull. For surgery, holes were drilled in the skull bilaterally as follows: for the PFC- anterior (A) from Bregma 3.7 mm, lateral (L) from midline 0.4mm; for the NAc A- 1.7 mm, L- 1.4 mm and 1.8 mm; for the CN posterior (P) from Bregma 0.2 mm, L- 2.0 mm and 3.0 mm; for the VTA P- 4.8 mm, L- 0.5 mm using the Paxinos and Watson “The Brain Atlas” [73] coordination. Bipolar stainless steel 80u electrode was used to make the non-specific electrolytic lesion with 2.0 mA DC current for 90 sec. at a depth of 3.2 mm and 4.2 mm; 1.0 mm and 1.6 mm; 4.0 mm and 5.2 mm and 8.2 mm and 8.5 mm within the PFC, NAc, CN and VTA respectively. For the chemical specific ablation of dopaminergic and glutamatergic neurons 8.0 ug 6OH DA (Sigma St. Louis, Mo, USA) dissolved in 2 uL of 0.9% Saline containing 0.2 mg/ml ascorbic acid and 4.0 uL of 1 ug/mL isotonic acid (Sigma St. Louis, Mo, USA) solution in depth similar to the non-specific electrolytic lesion for dopaminergic and the glutamatergic specific neurons ablation respectively. For the shame operation groups similar procedure (electrodes, cannula and coordination) were used but no current or injection were given [4,7,13-15,59,60,69]. The cannulas and the electrodes were then removed, and incision closed with wound staples.
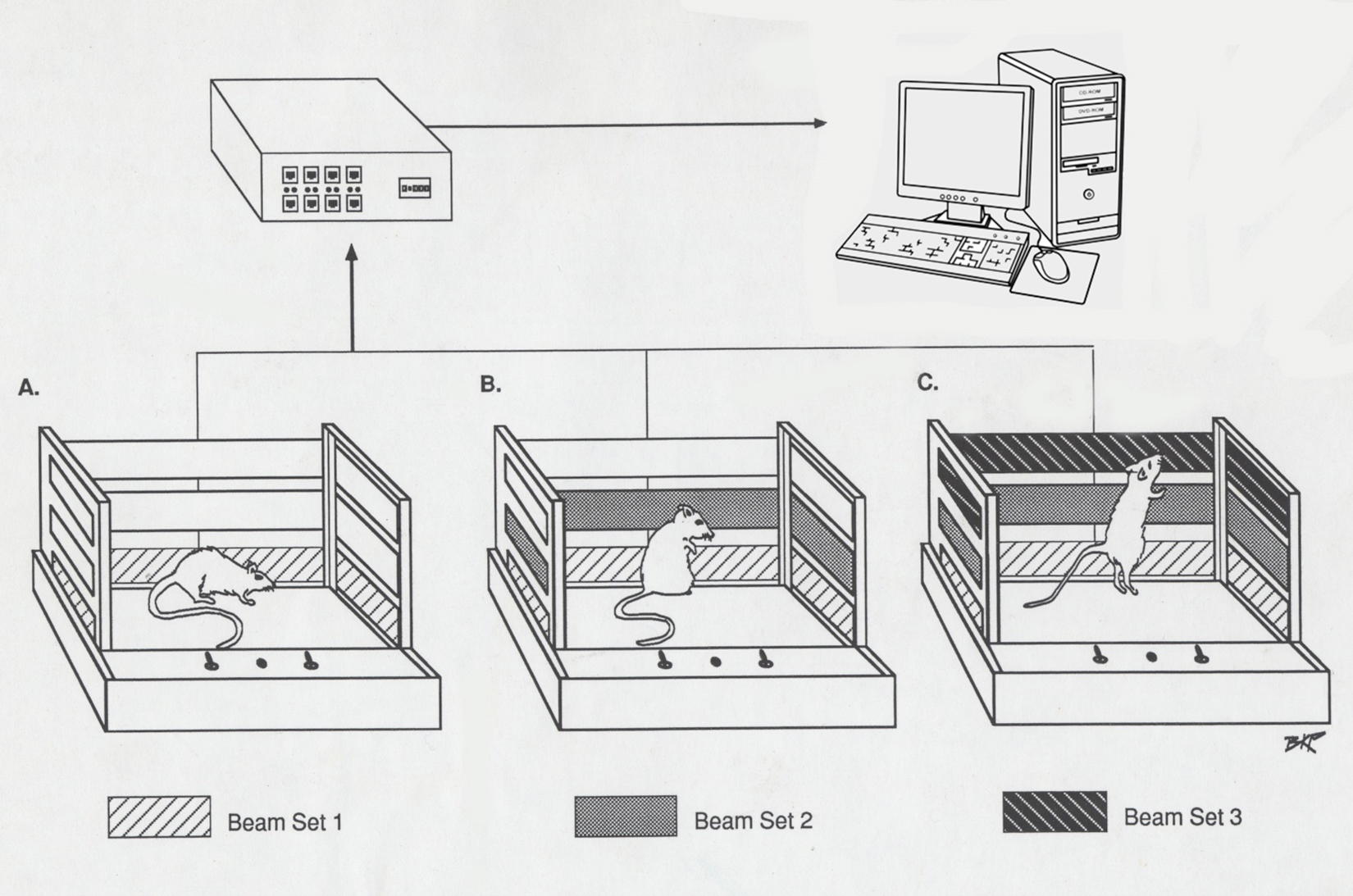
Figure 2: Figure 2 shows a schematic of the open field recording system used to monitor the animals and generate the indices of movement in the original reports [4,7,11,13-15,27,59].
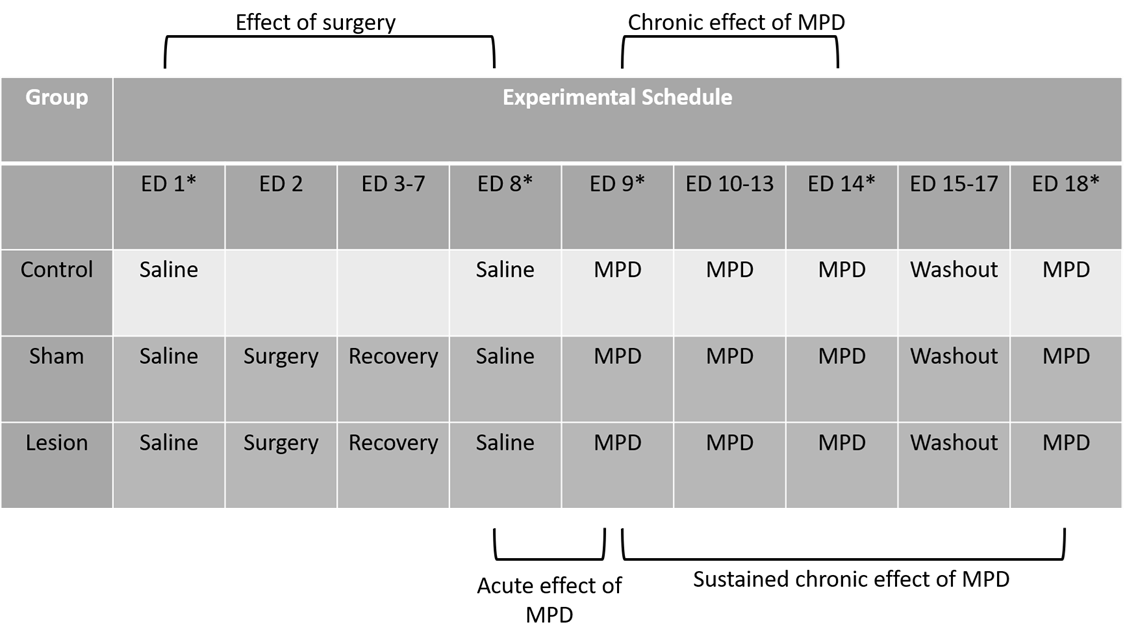
Table 1. Table 1 shows the experimental protocol of methylphenidate (MPD) administration and recording schedule. Displayed are the experimental days (ED’s) either normal saline or MPD was administered. * indicates day rats were behaviorally recorded post-injection. Lesion refers to the electrolytic, 6-OHDA toxin, or ibotenic acid toxin administered on ED 2. Brackets show the comparisons that were made: the post-surgical effect (ED 8 vs. ED 1), the acute effect of MPD (ED 9 vs. ED 8), and the chronic effect of MPD- sensitization- that is seen after sustained administration (ED 14 vs. ED 9) and persists after washout (ED 18 vs. ED 9). Full methodology can be found in the original reports [4,7,11,13-15,27,59].
Behavioral locomotive activity was recorded using the open field computerized animal activity monitoring system (CAAM, Accuscan Instruments, Inc., Columbus OH). The CAAM system consists of 3 arrays of 16 infrared light beams with sensors on the opposite side, spaced every 2.5 cm that cross orthogonally through the plexiglass cage (Figure 2). Movement of the rats interrupted the infrared light beams, and each beam-break detected by a sensor was collected as an event by the Accuscan Analyzer and transferred to a computer. Events over a 5-minute period were summed, giving 12 5-minute bins for each hour of observation. These bins were transferred to the OASIS data collecting software and three indices of behavioral locomotion were compiled for each collection period: Total movement (TM), total travelling distance (TD)- all forward locomotion in cm, horizontal activity (HA)- the overall movement in the lower level of the cage, and the number of stereotypic movements (NOS)- episodes of purposeless, repetitive movement in the upper level of the sensors separated by at least 1 second. Only HA, TD, & Nos were published, TM not. At the conclusion of the experiment, animals were overdosed with sodium pentobarbital and perfused with 10% formaldehyde. The brains were removed and stored in 10% formaldehyde. 60 µm thickness coronal sections were cut, stained, and scanned with a high-resolution scanner to identify lesion size and location correlated to the NAc using the Paximos and Watson rat brain atlas [73].
Rat behavioral locomotive activity was quantified by four compiled indices of movement (TM, HA, TD, NOS) obtained in twelve 5-minute bins collected the hour after injections for each rat were averaged across each experimental group based on the experimental day to allow for comparisons. Post-surgical manipulation effects on baseline behavioral locomotor activity were determined by comparing the animal’s activity after a saline injection before and after the surgical intervention (ED 8-Sal vs. ED 1-Sal). The acute effects of MPD were determined by comparing the first day of MPD administration to the post-surgical baseline (ED 9-MPD vs. ED 8-Sal). The effects of repetitive (chronic) MPD exposure over 6 consecutive days on behavioral locomotor activity were determined by comparing the final day of administration to the first, i.e. the induction phase (ED 14-MPD vs. ED 9-MPD). The effects of chronic MPD exposure following a washout period on behavioral locomotor activity were determined by comparing MPD re-challenge to the initial administration, i.e. the expression phase (ED 18-MPD vs. ED 9-MPD) (See Table 1). Significance of change among these within-group comparisons was determined by ANOVA, with repeated measures with adjustments for correlation among measurements within each animal. Post ad hoc comparisons were used to estimate changes between days within groups. A p-value<0.05 was considered statistically significant. The effects of each lesion were determined by comparing the treatment group to both the control and sham groups on each of the recording days (ED 1-Sal, ED 8-Sal, ED 9-MPD, ED 14-MPD, and ED 18-MPD). Significance of change among the between-group comparisons was determined with Turkey-Kramer Honest Significant Difference (HSD) post hoc test. A p-value<0.05 was considered statistically significant.
Result
The ventral tegmental area (Figure 3, VTA)
The VTA is a collection of primarily dopaminergic neurons distinct from the substantia nigra that gives rise to the dopaminergic projections of the mesocorticolimbic system which underlies the reward circuit and is critical for conditioned responses and chemical dependency [74-76]. Despite usually being considered a single entity, the VTA is quite heterogenous, and is comprised of four major divisions: the parafasciculus retroflex area (PFR), the ventral tegmental tail (VTT), the paranigral nucleus (PN), and the parabrachial pigmented area (PBP) [58]. The major outputs of the VTA are from the PN and PBP to the NAc and the PFC, which are critical for the initiation of reward functions via the mesolimbic system [19,58], and from the PFR and VTT to the diagonal band. The dopaminergic outflow of the VTA is thought to be modulated by its multiple afferent inputs, the most dominant being the glutamatergic input from the PFC [75]. Other inputs to the VTA originate from the NAc, the CN, the ventral pallidum, the rostromedial tegmental nucleus, the subthalamic nucleus, pedunculopontine tegmental and laterodorsal tegmental nuclei, the bed nucleus of the stria terminalis, and the superior colliculus [17,18,40,41,77-81].
Both glutaminergic and dopaminergic signaling in the VTA have been shown to be critical for the animal response to MPD. Specific glutaminergic ablation of the VTA abolishes any response to MPD acutely or chronically, indicating that glutaminergic signaling with the VTA is critical for the acute and chronic behavioral responses to MPD (Figure 3) [59]. Dopaminergic ablation however only prevents the acute response to MPD, the chronic response of behavioral sensitization is left intact (Figure 3) [59]. Significant attention has been paid to the functions of the VTA’s dopamine projections, which have been shown to initiate the mesocorticolimbic reward system [49,81-83], however this finding of glutamate in the VTA being critical is novel and presents a further nuance to the complexities of this nucleus. Notably, glutaminergic ablation of the prefrontal cortex also precludes behavioral sensitization [84,85], which agrees with the hypothesis that the PFC/VTA is the predominant pathway to regulating behavioral sensitization.
There is significant evidence that differing sections of the VTA induce different behavioral outcomes. The medial VTA is comprised of an inhibitory pathway that negatively regulates behavioral activities [14,81,86-88] while the lateral VTA appears to mediate reward functions such as behavioral augmentation [19,20,76,77]. Excitatory D1 receptors appear to be the predominant type in the lateral reward pathway [89], with inhibitory D2 making up the majority of the medial inhibitory pathway receptor type [90,91]. The functional and anatomic distinction with in the VTA would appear to explain the results of non-specific electrolytic ablation of the VTA causing an acute increase in behavioral activity with no change to the overall response to MPD administration (Figure 3) [59]. Ablation of the medial inhibitory VTA with an intact lateral reward VTA would produce larger behavioral increases while maintaining the response to MPD.
Identification and targeting of the VTA’s subregions (medial vs. lateral, PFR vs. VTT vs. PN vs. PBP) has proved to be exceedingly difficult, even using in vivo electrophysiologic studies [92]. These studies of the heterogeneity of the VTA appear to disagree with the classical view of the VTA solely as the dopaminergic origin of the mesolimbic and mesocortical pathways, and hints at the higher order modulation required to produce complex behavioral responses. Further work using in vivo recording, lesions, and microinjections investigating the subdivisions of the VTA represent future avenues for understanding this nucleus.
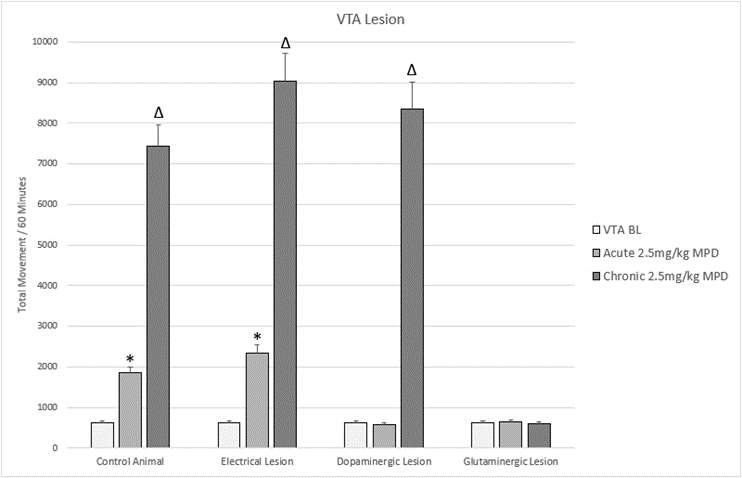
Figure 3. Figure 3 shows a representation of animal total movement (TM) following acute and chronic methylphenidate (MPD) in animals that were subjected to lesions to the VTA.
* = indicate significant p<0.05 different from experimental day 1 (ED 1) baseline (BL),
(ED1 MPD/ ED1 BL); Δ = indicate significant p<0.05 different from experimental day 1 (ED 1) methylphenidate (MPD) 2.5mg/kg, i.e., (ED10 MPD/ ED1 MPD).
The nucleus accumbens (Figure 4, NAc)
The nucleus accumbens (NAc) is critical for motivation, emotion, limbic functions, and motor execution. It is composed predominantly of dopaminergic median spiny neuron’s (MSN’s) and is divided into a shell and core that are anatomically and functionally distinct [93-95]. The NAc shell seems to mediate reward and addiction behaviors, while the core appears to modulate conditioned response and spatial learning [94,96,97]. The primary input to the NAc is from the VTA, but it also receives input from the substantia nigra, the amygdala, the hippocampus, and the PFC. The output from the NAc is via its MSN’s to various basal ganglia and midbrain structures including (but not limited to) the substantia nigra, the VTA, the ventral pallidum, the thalamus, the globus pallidus, the subpallidus, and the stria terminalis [94,98,99].
Changes in accumbal dopamine have been shown to be critical for reward circuit responses. Psychostimulants such as MPD have been shown to cause an increase in dopaminergic transmission from the VTA to the NAc, and increased dopamine within the NAc has also been shown to lead to increased locomotion [100-102]. It has been consistently reported that direct chronic microinjection of other dopaminergic agonists such as amphetamine, cocaine, or morphine into the NAc can induce behavioral sensitization [103-105], suggesting that the NAc is involved in the induction of behavioral sensitization. Psychostimulant exposure increases dendritic branch points and spines of the NAc’s MSN’s, and MPD has been shown to cause increased spine formation in excitatory MSN-D1 dopamine receptors but not inhibitory MSN-D2 dopamine receptor spines [106,107]. This excitatory effect of psychostimulants is greatest in the shell [108-110], indicating this component of the NAc is critical to mediating chronic psychostimulant effects.
Lesions to the NAc have confirmed its role in responding to acute and chronic MPD. Non-specific electrolytic lesions to the NAc have resulted in an exaggerated acute response to MPD and a loss of behavioral sensitization with chronic exposure (Figure 4) [13]. This indicates that in addition to its role in the response to MPD, the NAc serves as an inhibitor of behavioral activity [111]. The NAc’s inhibitory role in behavioral activity can be seen following treatment with dopamine modulators that attenuate activity [112,113], and could serve as an autoregulatory system to control excessive dopaminergic signaling.
Selective lesions to the dopaminergic signaling of the NAc also resulted in a loss of behavioral sensitization to cocaine, amphetamine, and MPD (Figure 4) [15,114,115]. One study showed two distinct responses in rats following dopaminergic lesions of the NAc. While some animals exhibited no increase in locomotor activity after the acute injection of MPD, others showed a significantly elevated locomotor activity following MPD and this difference persisted throughout the length of the study [15]. Those animals that showed an increase in behavioral activity following acute MPD did not develop behavioral sensitization, i.e. a further increase beyond the acute effect, while those that showed no behavioral change following the dopaminergic lesion did show behavioral sensitization following repetitive (chronic) MPD exposure [15]. This work seems to verify that NAc dopamine is a critical component of the behavioral response both acutely and chronically to MPD, but the heterogeneity of responses raises more questions than it answers. The author notes that the accuracy of the lesion (core, shell or both) is unknown as the lesions could not be verified independently and therefore it is possible that the difference in acute response to psychostimulant administration may be due to differences in the size or location of the lesion [15].
The glutaminergic signaling of the NAc appears to also modulate the effect of MPD. Selective lesions to the glutaminergic NAc signaling system do not lead to a gross disturbance in the acute or chronic response to MPD (Figure 4) [60], however when broken down into different locomotor expression of movement (horizontal activity vs. total distance vs. stereotypic movements), a significant difference is seen between groups [60]. Specifically, more goal directed forward movement as measured by horizontal activity (HA) was attenuated while stereotypic activity as measured by the number of stereotypic movements (NOS) was augmented following glutaminergic ablation of the NAc; total distance (TD) traveling remained unchanged [60]. This work seems to indicate that it is volitional movement that is modulated by glutaminergic signaling in the NAc, which fits with glutaminergic input from the PFC being critical to movement [116-120], and with a more global picture of glutaminergic signaling being critical to modulating the motor outcomes of the rewards circuit [4,7,19,21,70,87].
However, this lesion was centered in the NAc core and did not differentiate from the core versus the shell. The specific attenuation of volitional movement corroborates with other work showing that in addition to the shell/core distinction, different pathways within the NAc govern motivation and behavioral actions [121,122]. And while the NAc shell was initially thought to govern the response to psychostimulants, the NAc core has also been shown to participate in the response as well [109,123,124], further reinforcing the evidence that the anatomic distinctions are really secondary to the signaling pathways within the NAc. This all hint at the differing roles of the NAc core, shell, and the circuits between them.
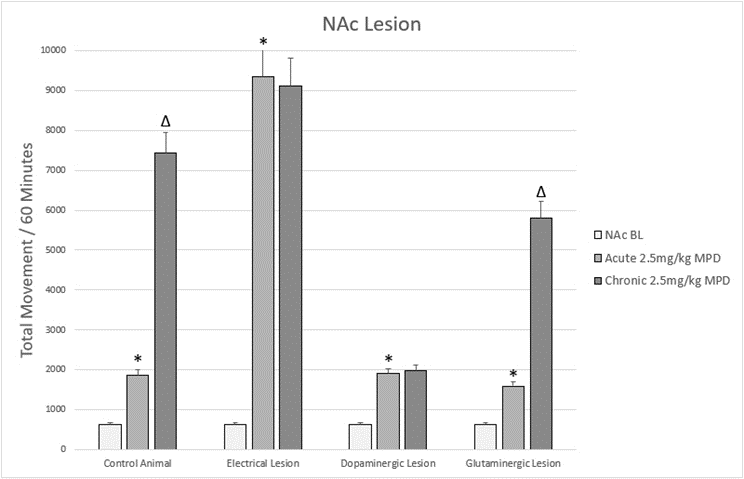
Figure 4. Figure 4 shows a representation of animal TM following acute and chronic methylphenidate (MPD) in animals that were subjected to lesions to the NAc.
* = indicate significant p<0.05 different from experimental day 1 (ED 1) baseline (BL), (ED1 MPD/ ED1 BL); Δ = indicate significant p<0.05 different from experimental day 1 (ED 1) methylphenidate (MPD) 2.5mg/kg, i.e., (ED10 MPD/ ED1 MPD).
The caudate nucleus (Figure 5, CN)
The caudate nucleus (CN) is unique in that it belongs to both the extrapyramidal motor system and reward/motive circuit [19,20,125,126]. In rats, who lack an internal capsule, the caudate is blended with the putamen forming the caudoputamen or dorsal striatum which can similarly be divided into a medial and lateral component. The CN is comprised primarily of catecholaminergic dopaminergic medium spiny neurons (MSNs) that were originally thought to be inhibitory regulators of movement [127,128], however further work showed its functions to more complex [129]. The MSN’s of the CN express excitatory D1-dopamine receptors and inhibitory D2-dopamine receptors [130,131]. These neurons project via the direct pathway, the ansa leticularis, and the indirect pathway, the lenticular fasiculus, [125] to modulate movement. The indirect pathway expresses primarily inhibitory D2-dopamine receptors and exerts its effects via the globus pallidus externa and subthalamus. The direct pathway primarily expresses stimulatory D1 receptors to inhibit the globus pallidus interna. From there, both pathways project to the motor nuclei of the thalamus then to the cortex [125,130,132,133]. The CN receives input from other reward circuit nuclei such as the VTA, the NAc, and the PFC which assist in mediating behavioral sensitization following chronic psychostimulant administration [13-15,19,-21,134,135].
The dopaminergic and glutaminergic signal pathways of the CN have been shown to be critical for the effects of MPD. Increased dopamine transmission in the CN in response to psychostimulant exposure contributes to the increased locomotive activity that is characteristic of psychostimulants (Figure 5) [4,7,8,136]. Specific lesions to the dopaminergic neurons of the CN extinguishes any response to MPD both acutely and chronically, and specific antagonism of the D1 receptor can prevent the motor response to MPD, indicating that the excitatory pathway is the primary target of MPD (Figure 5) [4,7,136,137]. Glutamate signaling regulates the output of the MSN’s to produce the chronic effects of MPD as shown by ablation of glutaminergic signaling within the CN that preventing the chronic effect of MPD such as behavioral sensitization, but not the acute effect (Figure 5) [4]. Co-localized glutamate and dopamine receptors on a subset of MSN’s and an alteration of dopamine synthesis capacity in response to local CN glutamate could explain the modulatory role of glutaminergic signaling, however more work would need to done to verify this [135,138-141].
The CN has been shown to be critical for learning and memory, mediated by several pathways. Several studies have shown that animal memory is enhanced by increased CN dopamine and impaired by dopaminergic lesions [142,143], consistent with the theory of dopamine-mediated memory consolidation. However, glutaminergic signaling in the CN has also been shown to participate in long-term learning as well. Glutamine infusion into the CN has been shown to strengthen learned behavior, and N-methyl-D-aspartate (NMDA) receptors, a subtype of glutamate receptors, in the CN are required for operant learning in rats [144,145]. Additionally, non-specific systemic glutamate antagonists can reduce the stereotyped behavioral responses to psychoactive substances [146,147], and although this work is not CN specific it lends credence to a glutaminergic component to learning. The lack of behavioral sensitization to MPD after specific glutaminergic ablation supports glutaminergic signaling within the CN as mediator of long-term learning and substance abuse associated with chronic MPD use [4]. These findings seem to indicate a concomitant neuromodulatory role of CN glutamate and dopamine for learned behaviors an individual exhibit in association with chronic psychostimulant use. And while evidence exists for some anatomic and function divisions between the medial and lateral CN in cognitive and association functions respectively [143], no work has been targeted enough to examine if the dopaminergic and glutaminergic mediated learning pathways are similarly separated.
Non-specific electrolytic lesions to the CN have failed to affect the acute or chronic response to MPD (Figure 5) indicating that current electrolytic lesion targeting both afferent, the direct excitatory and the indirect inhibitory circuit and supports the CN as a heterogenous structure in both form and function [4,7].
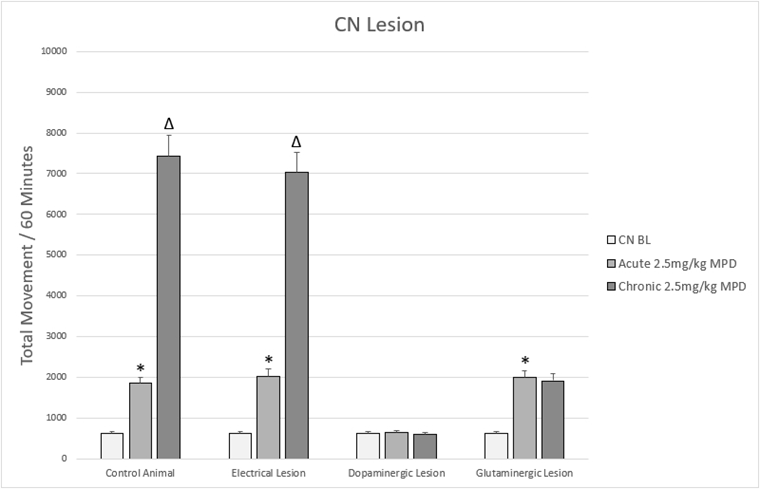
Figure 5. Figure 5 shows a representation of animal TM following acute and chronic methylphenidate (MPD) in animals that were subjected to lesions to the CN.
* = indicate significant p<0.05 different from experimental day 1 (ED 1) baseline (BL), (ED1 MPD/ ED1 BL); Δ = indicate significant p<0.05 different from experimental day 1 (ED 1) methylphenidate (MPD) 2.5mg/kg, i.e., (ED10 MPD/ ED1 MPD).
The prefrontal cortex (Figure 6, PFC)
The prefrontal cortex (PFC) is a large territory of tissue at the anterior pole of the brain. It is critical to a diverse range of cognitive functions such as emotion, conscious decisions, and memory [148]. The PFC serves as a significant source of the excitatory amino acid glutamate, which it projects to the VTA and NAc to modulate dopaminergic signaling at these nuclei [48,49,149-151].
The PFC is critical in mediating behavioral sensitization to psychostimulants. Non-specific ablation of the rat PFC as well as dopaminergic specific lesion did not prevent the MPD acute effect while, prevent the MPD repetitive (chronic) effect eliciting behavioral sensitization in response to chronic MPD administration (Figure 6) [11,69]. However, glutaminergic specific ablation has been shown to prevent MPD-induced hyperactivity acutely, however the chronic response of sensitization is maintained and even exaggerated (Figure 6) [14]. These differing responses to non-specific electrolytic lesions as compared to a selective chemical lesion is likely due to the numerous different neuronal pathways affected by a non-selective lesion [11,14]. They are consistent with the known glutamatergic efferents to the VTA and NAc. Glutamate from the PFC excites VTA dopaminergic neurons, which increases dopamine release in the NAc [118-120]. Increased dopamine within the NAc disinhibits motor inhibition, thus leading to increased locomotion [151,152]. This finding has been replicated with other psychostimulants, further implicating glutamate from the PFC as a key component of the neuroadaptive response to psychostimulants [80,151,153-157]. The enhanced chronic response following MPD in the setting of glutaminergic specific ablation is likely due to the persistence of non-glutaminergic pathways that are uncovered following the specific lesion but destroyed in the non-specific lesion, and further indicates the diverse neuronal populations in the PFC [11,14]
The PFC cytoarchitecture however is highly heterogeneous, containing norepinephrine, dopamine, α2 adrenoreceptors, and GABA in addition to its primary glutaminergic neurons [158-160]. There are two subgroups of DA receptors: excitatory D1 and inhibitory D2 receptors, with D1 DA receptors are expressed in a higher density compared to D2 DA in the PFC [161,162]. These receptors have been shown to modulate the excitability of PFC neurons, with D1 receptor activation being able to directly excite PFC neurons [162-164]. One study has shown that specific dopaminergic ablation of the PFC did not alter the acute behavioral change to MPD, but did prevent behavioral sensitization following chronic MPD (Figure 6) [15]. However, reports of dopaminergic lesions to the PFC are mixed: with some reporting behavioral changes consistent with the prior study [114,165,166], while others report inconsistent motor activity changes following the lesion [166,167]. While these differences could certainly have been due to methodology, all the reports indicate that PFC dopamine plays a role in regulating both the motor response to acute MPD and the neuroadaptation to chronic MPD characteristic of the expression of behavioral sensitization.
However, rodent studies of the PFC should be considered with the recognition that they are poor surrogates for the human counterpart. The human PFC contains multiple divisions, of which only a few share homologies with a rodent counterpart [148,168]. Indeed, this is readily apparent from the lack of higher order social, emotional, and cognitive functions in rodents. So, while these studies of pathways and signaling in the rodent are not directly translatable to man, they provide an important understanding of the primitive pathways that drive complex neurocognitive functions such as behavioral sensitization and substance abuse. Further work in this region promises to deliver even more complete understanding of the basis of volitional and non-volitional motivation.
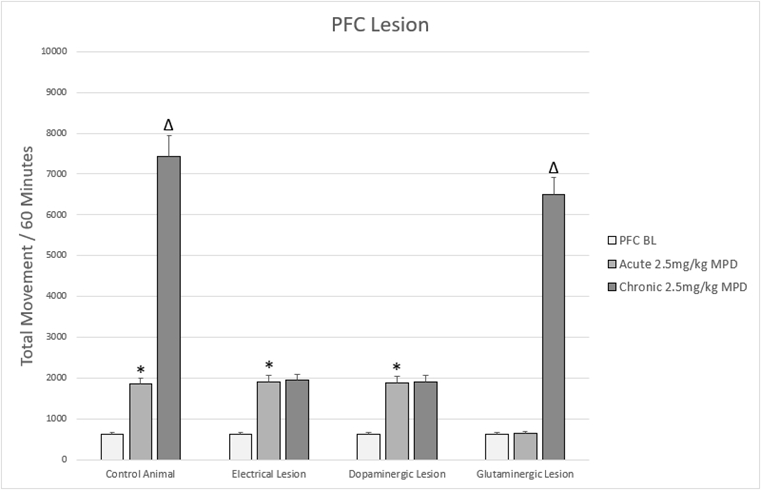
Figure 6: Figure 6 shows a representation of animal TM following acute and chronic methylphenidate (MPD) in animals that were subjected to lesions to the PFC.
* = indicate significant p<0.05 different from experimental day 1 (ED 1) baseline (BL), (ED1 MPD/ ED1 BL); Δ = indicate significant p<0.05 different from experimental day 1 (ED 1) methylphenidate (MPD) 2.5mg/kg, i.e., (ED10 MPD/ ED1 MPD).
Conclusions
The NAc, VTA, CN, and PFC have been shown to be critical in the acute and chronic response to MPD. Through specific and non-specific lesions (Figures 3-6), the functions of the nuclei and their composite signaling pathways is better understood. But as more is learned, further questions are raised regarding anatomic and functional distinctions of the various subdivisions noted. Further refinement of in vivo identification and targeting of these subdivisions will allow for a more accurate understanding of the reward circuit and its response to psychostimulants such as MPD.
Acknowledgements
The authors would like to acknowledge Alonso Carrasco, Catherine Claussen, Samuel Chong, Samuel Floren, Natasha Kharas, Min Lee, Adam Podet, Alan Swann, Ming Thomas, and Sheshali Wanchoo who contributed to the studies. These studies were supported by the National Institute of Health NIH RO 1 DA- 027220.
Declarations
Funding and disclosure
The authors have no relevant disclosures.
Conflict of interest
There are no conflicts of interest among any of the listed authors.
Consent for publication
All authors have approved the manuscript and agree with its submission.
References
2. Greely H, Sahakian B, Harris J, Kessler RC, Gazzaniga M, Campbell P, et al. Towards responsible use of cognitive-enhancing drugs by the healthy. Nature. 2008 Dec 11;456(7223):702-5.
3. Jain R, Chang CC, Koto M, Geldenhuys A, Nichol R, Joubert G. Non-medical use of methylphenidate among medical students of the University of the Free State. South African Journal of Psychiatry. 2017 Jan 25;23.
4. King N, Floren S, Kharas N, Thomas M, Dafny N. Glutaminergic signaling in the caudate nucleus is required for behavioral sensitization to methylphenidate. Pharmacology, Biochemistry and Behavior. 2019;184:172737.
5. Pauly V, Frauger E, Lepelley M, Mallaret M, Boucherie Q, Micallef J. Patterns and profiles of methylphenidate use both in children and adults. British Journal of Clinical Pharmacology. 2018 Jun;84(6):1215-27.
6. Wilens TE, Adler LA, Adams J, Sgambati S, Rotrosen J, Sawtelle R, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. Journal of the American Academy of Child & Adolescent Psychiatry. 2008 Jan 1;47(1):21-31.
7. Claussen CM, Chong SL, Dafny N. Selective bilateral lesion to caudate nucleus modulates the acute and chronic methylphenidate effects. Pharmacology Biochemistry and Behavior. 2012 Apr 1;101(2):208-16.
8. Claussen CM, Witte LJ, Dafny N. Single exposure of dopamine D1 antagonist prevents and D2 antagonist attenuates methylphenidate effect. Journal of Experimental Pharmacology. 2015 Apr 20:1-9.
9. Gaytan O, al-Rahim S, Swann A, Dafny N. Sensitization to locomotor effects of methylphenidate in the rat. Life Sciences. 1997 Jul 18;61(8):PL101-7.
10. Gaytan O, Sripada S, Swann A, Dafny N. Blockade of sensitization to methylphenidate by MK-801: partial dissociation from motor effects. Neuropharmacology. 2001 Jan 1;40(2):298-309.
11. Lee MJ, Swann AC, Dafny N. Methylphenidate sensitization is prevented by prefrontal cortex lesion. Brain Research Bulletin. 2008 May 15;76(1-2):131-40.
12. Lee MJ, Yang PB, Wilcox VT, Burau KD, Swann AC, Dafny N. Does repetitive Ritalin injection produce long-term effects on SD female adolescent rats?. Neuropharmacology. 2009;3(57):201-7.
13. Podet A, Lee MJ, Swann AC, Dafny N. Nucleus accumbens lesions modulate the effects of methylphenidate. Brain Research Bulletin. 2010 Jul 30;82(5-6):293-301.
14. Wanchoo SJ, Swann AC, Dafny N. Descending glutamatergic pathways of PFC are involved in acute and chronic action of methylphenidate. Brain Research. 2009 Nov 16;1301:68-79.
15. Wanchoo SJ, Lee MJ, Swann AC, Dafny N. Bilateral six-hydroxydopamine administration to PFC prevents the expression of behavioral sensitization to methylphenidate. Brain Research. 2010 Feb 2;1312:89-100.
16. Yang P, Singhal N, Modi G, Swann A, Dafny N. Effects of lithium chloride on induction and expression of methylphenidate sensitization. European Journal of Pharmacology. 2001 Aug 24;426(1-2):65-72.
17. Yang PB, Amini B, Swann AC, Dafny N. Strain differences in the behavioral responses of male rats to chronically administered methylphenidate. Brain Research. 2003 May 9;971(2):139-52.
18. Yang PB, Swann AC, Dafny N. Chronic pretreatment with methylphenidate induces cross-sensitization with amphetamine. Life Sciences. 2003 Oct 17;73(22):2899-911.
19. Yang PB, Swann AC, Dafny N. Sensory-evoked potentials recordings from the ventral tegmental area, nucleus accumbens, prefrontal cortex, and caudate nucleus and locomotor activity are modulated in dose–response characteristics by methylphenidate. Brain Research. 2006 Feb 16;1073:164-74.
20. Yang PB, Swann AC, Dafny N. Chronic methylphenidate modulates locomotor activity and sensory evoked responses in the VTA and NAc of freely behaving rats. Neuropharmacology. 2006 Sep 1;51(3):546-56.
21. Yang PB, Swann AC, Dafny N. Chronic administration of methylphenidate produces neurophysiological and behavioral sensitization. Brain Research. 2007 May 11;1145:66-80.
22. Yang PB, Swann AC, Dafny N. Methylphenidate treated at the test cage–dose-dependent sensitization or tolerance depend on the behavioral assay used. Crit Rev Neurobiol. 2007;19(1):59-77.
23. Yang PB, Cuellar III DO, Swann AC, Dafny N. Age and genetic strain differences in response to chronic methylphenidate administration. Behavioural Brain Research. 2011 Mar 17;218(1):206-17.
24. Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine: comparison with amphetamine. Journal of Neurochemistry. 1997 May;68(5):2032-7.
25. Medina AC, Kabani A, Reyes-Vasquez C, Dafny N. Age differences to methylphenidate-NAc neuronal and behavioral recordings from freely behaving animals. Journal of Neural Transmission. 2022 Aug;129(8):1061-76.
26. Venkataraman SS, Joseph M, Dafny N. Concomitant behavioral and prefrontal cortex neuronal responses following acute and chronic methylphenidate exposure in adolescent and adult rats. Brain Research Bulletin. 2019 Jan 1;144:200-12.
27. Venkataraman SS, Claussen CM, Kharas N, Dafny N. The prefrontal cortex and the caudate nucleus respond conjointly to methylphenidate (Ritalin). Concomitant behavioral and neuronal recording study. Brain Research Bulletin. 2020 Apr 1;157:77-89.
28. Teo SK, Stirling DI, Hoberman AM, Christian MS, Thomas SD, Khetani VD. D‐methylphenidate and D, L‐methylphenidate are not developmental toxicants in rats and rabbits. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 2003 Apr;68(2):162-71.
29. Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley JS, et al. Is methylphenidate like cocaine?: Studies on their pharmacokinetics and distribution in the human brain. Archives of General Psychiatry. 1995 Jun 1;52(6):456-63.
30. Volkow ND, Wang GJ, Fowler JS, Fischman M, Foltin R, Abumrad NN, et al. Methylphenidate and cocaine have a similar in vivo potency to block dopamine transporters in the human brain. Life Sciences. 1999 May 28;65(1):PL7-12.
31. Challman TD, Lipsky JJ. Methylphenidate: its pharmacology and uses. Mayo Clinic Proceedings. 2000 Jul 1; 75(7):711-721.
32. Crutchley A, Temlett JA. Methylphenidate (Ritalin) use and abuse. South African Medical Journal. 1999;89(10):1076-9.
33. Wilens TE, Biederman J, Mick E, Faraone SV, Spencer T. Attention deficit hyperactivity disorder (ADHD) is associated with early onset substance use disorders. The Journal of Nervous and Mental Disease. 1997 Aug 1;185(8):475-82.
34. Gatley SJ, Volkow ND, Gifford AN, Fowler JS, Dewey SL, Ding YS, et al. Dopamine-transporter occupancy after intravenous doses of cocaine and methylphenidate in mice and humans. Psychopharmacology. 1999 Sep;146:93-100.
35. John CE, Jones SR. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudate-putamen and substantia nigra slices. Neuropharmacology. 2007 Jun;52(8):1596-605.
36. Corrigall WA, Franklin KB, Coen KM, Clarke PB. The mesolimbic dopaminergic system is implicated in the reinforcing effects of nicotine. Psychopharmacology. 1992 Jun;107:285-9.
37. Pierce RC, Kumaresan V. The mesolimbic dopamine system: the final common pathway for the reinforcing effect of drugs of abuse?. Neuroscience & Biobehavioral Reviews. 2006 Jan 1;30(2):215-38.
38. Amini B, Yang PB, Swann AC, Dafny N. Differential locomotor responses in male rats from three strains to acute methylphenidate. International Journal of Neuroscience. 2004 Jan 1;114(9):1063-84.
39. Brandon CL, Marinelli M, Baker LK, White FJ. Enhanced reactivity and vulnerability to cocaine following methylphenidate treatment in adolescent rats. Neuropsychopharmacology. 2001 Nov 1;25(5):651-61.
40. Gaytan O, Ghelani D, Martin S, Swann A, Dafny N. Dose response characteristics of methylphenidate on different indices of rats' locomotor activity at the beginning of the dark cycle. Brain Research. 1996 Jul 15;727(1-2):13-21.
41. Gaytan O, Nason R, Alagugurusamy R, Swann A, Dafny N. MK-801 blocks the development of sensitization to the locomotor effects of methylphenidate. Brain Res Bull. 2000 Apr;51(6):485-92.
42. Kollins SH, English J, Robinson R, Hallyburton M, Chrisman AK. Reinforcing and subjective effects of methylphenidate in adults with and without attention deficit hyperactivity disorder (ADHD). Psychopharmacology (Berl). 2009 May;204(1):73-83.
43. Solanto MV. Neuropsychopharmacological mechanisms of stimulant drug action in attention-deficit hyperactivity disorder: a review and integration. Behavioural Brain Research. 1998 Jul 1;94(1):127-52.
44. Berridge KC, Robinson TE. The mind of an addicted brain: Neural sensitization of wanting versus liking. Current Directions in Psychological Science. 1995 Jun;4(3):71-6.
45. Crawford CA, McDougall SA, Meier TL, Collins RL, Watson JB. Repeated methylphenidate treatment induces behavioral sensitization and decreases protein kinase A and dopamine-stimulated adenylyl cyclase activity in the dorsal striatum. Psychopharmacology. 1998 Feb;136:34-43.
46. Dafny N, Reyes-Vasquez C, Liu Y. The serotonergic signaling and the dorsal raphe (DR) neurons in adolescent rats are the most engaging in response to acute and chronic methylphenidate as compare to other neuronal activities recorded from other five brain nuclei. Journal of Clinical Pharmacology and Therapeutics. 2022;3:8-21.
47. Kalivas PW. Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug and Alcohol Dependence. 1995 Feb 1;37(2):95-100.
48. Kalivas PW, Churchill L, Klitenick MA. The circuitry mediating the translation of motivational stimuli into adaptive motor responses. In: Kalivas PW, Barnes CD, (Eds). Limbic Motor Circuits and Neuropsychiatry. Boca Raton: CRC Press. 1993 Jun 4; pp. 237-288.
49. Kalivas PW, Sorg BA, Hooks MS. The pharmacology and neural circuitry of sensitization to psychostimulants. Behavioural Pharmacology. 1993 Aug 1;4(4):315-34.
50. Dafny N, Yang PB. The role of age, genotype, sex, and route of acute and chronic administration of methylphenidate: a review of its locomotor effects. Brain Research bulletin. 2006 Feb 15;68(6):393-405.
51. Berridge KC, Kringelbach ML. Affective neuroscience of pleasure: reward in humans and animals. Psychopharmacology. 2008 Aug;199:457-80.
52. Gardner EL. Brain reward mechanisms. Substance abuse: A comprehensive textbook. 1997:51-85.
53. Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010 Jan;35(1):4-26.
54. Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nature Reviews Neuroscience. 2013 Sep;14(9):609-25.
55. Anderson-Mooney AJ, Dodd JN, Scott A, Guller L. The nervous system and addictions: essentials for clinicians. In: Neuropathology of Drug Addictions and Substance Misuse. Academic Press. 2016 Jan 1; pp. 3-13.
56. Haber SN. Anatomy and Connectivity of the Reward Circuit. InDecision Neuroscience 2017 Jan 1 (pp. 3-19). Academic Press.
57. Ledonne A, Mercuri NB. Current concepts on the physiopathological relevance of dopaminergic receptors. Frontiers In Cellular Neuroscience. 2017 Feb 8;11:27.
58. Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens–olfactory tubercle complex. BrainResearch Reviews. 2007 Nov 1;56(1):27-78.
59. Floren S, King N, Carrasco A, Dafny N. Glutamate and dopamine in the VTA participate differently in the acute and chronic effect of methylphenidate. Behavioural Brain Research. 2020 Feb 17;380:112390.
60. Ming T, Dafny N. Glutaminergic Signaling in the Nucleus Accumbens Modulates the Behavioral Response to Acute and Chronic Methylphenidate. Journal of Experimental Neurology. 2021;2(1):49-61.
61. Blum D, Torch S, Lambeng N, Nissou MF, Benabid AL, Sadoul R, et al. Molecular pathways involved in the neurotoxicity of 6-OHDA, dopamine and MPTP: contribution to the apoptotic theory in Parkinson's disease. Progress in Neurobiology. 2001 Oct 1;65(2):135-72.
62. Iversen L. 1973. Actions of 6-hydroxydopamine on catecholamine-containing neurons in the central nervous system. Advances in Neurology. 3:243-255.
63. Luthman J, Fredriksson A, Sundström E, Jonsson G, Archer T. Selective lesion of central dopamine or noradrenaline neuron systems in the neonatal rat: motor behavior and monoamine alterations at adult stage. Behavioural Brain Research. 1989 Jul 1;33(3):267-77.
64. Kouvelas ED, Mitsacos A, Angelatou F, Hatziefthimiou A, Tsiotos P, Voukelatou G. Glutamate Receptors in Mammalian Cerebellum: Alterations in Human Ataxic Disorders and Cerebellar Mutant Mice. In: Plaitakis A (eds) Cerebellar Degenerations: Clinical Neurobiology. Foundations of Neurology. vol 2, Springer: Boston, MA. 1992.
65. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. The Journal of Nutrition. 2000 Apr 1;130(4):1007S-15S.
66. Schwarcz R, Hökfelt T, Fuxe K, Jonsson G, Goldstein M, Terenius L. Ibotenic acid-induced neuronal degeneration: a morphological and neurochemical study. Experimental Brain Research. 1979 Oct;37:199-216.
67. Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Research Reviews. 2004 Jul 1;45(3):250-65.
68. Ihezie SA, Thomas MM, Dafny N. Acute and chronic methylphenidate administration in intact and VTA-specific and nonspecific lesioned rats. J Neural Transm (Vienna). 2019 Feb;126(2):173-182.
69. Tang A, Wanchoo SJ, Swann AC, Dafny N. Psychostimulant treatment for ADHD is modulated by prefrontal cortex manipulation. Brain research bulletin. 2009 Dec 16;80(6):353-358.
70. Jones Z, Dafny N. Acute and chronic dose–response effect of methylphenidate on ventral tegmental area neurons correlated with animal behavior. Journal of Neural Transmission. 2014 Mar;121:327-45.
71. Karim TJ, Reyes-Vazquez C, Dafny N. Comparison of the VTA and LC response to methylphenidate: a concomitant behavioral and neuronal study of adolescent male rats. Journal of Neurophysiology. 2017 Sep 1;118(3):1501-14.
72. Karim TJ, Aksel C, Kharas N, Reyes-Vasquez C, Dafny N. Caudate nucleus neurons participate in methylphenidate function: Behavioral and neuronal recordings from freely behaving adolescent rats. Brain Res Bull. 2018 Sep;142:241-252.
73. Paxinos G, Watson C. The rat brain in stereotaxic coordinates. 2nd Edition. Academic Press; 1986.
74. Charara A, Smith Y, Parent A. Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris‐leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. Journal of Comparative Neurology. 1996 Jan 8;364(2):254-66.
75. Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011 Jun 9;70(5):855-62.
76. Morikawa H, Paladini CA. Dynamic regulation of midbrain dopamine neuron activity: intrinsic, synaptic, and plasticity mechanisms. Neuroscience. 2011 Dec 15;198:95-111.
77. Braun AR, Jaskiw GE, Vladar K, Sexton RH, Kolachana BS, Weinberger DR. Effects of ibotenic acid lesion of the medial prefrontal cortex on dopamine agonist-related behaviors in the rat. Pharmacology Biochemistry and Behavior. 1993 Sep 1;46(1):51-60.
78. Jackson EA, Neumeyer JL, Kelly PH. Behavioral activity of some novel aporphines in rats with 6-hydroxydopamine lesions of caudate or nucleus accumbens. European Journal of Pharmacology. 1983 Jan 28;87(1):15-23.
79. Jaskiw GE, Karoum F, Freed WJ, Phillips I, Kleinman JE, Weinberger DR. Effect of ibotenic acid lesions of the medial prefrontal cortex on amphetamine-induced locomotion and regional brain catecholamine concentrations in the rat. Brain Research. 1990 Nov 26;534(1-2):263-72.
80. Li Y, Hu XT, Berney TG, Vartanian AJ, Stine CD, Wolf ME, White et al. Both glutamate receptor antagonists and prefrontal cortex lesions prevent induction of cocaine sensitization and associated neuroadaptations. Synapse. 1999 Dec 1;34(3):169-80.
81. Watabe-Uchida M, Zhu L, Ogawa SK, Vamanrao A, Uchida N. Whole-brain mapping of direct inputs to midbrain dopamine neurons. Neuron. 2012 Jun 7;74(5):858-73.
82. Salamone JD, Correa M. The mysterious motivational functions of mesolimbic dopamine. Neuron. 2012 Nov 8;76(3):470-85.
83. Sellings LH, Clarke PB. 6‐Hydroxydopamine lesions of nucleus accumbens core abolish amphetamine‐induced-conditioned activity. Synapse. 2006 May;59(6):374-7.
84. Hemby SE, Horman B, Tang W. Differential regulation of ionotropic glutamate receptor subunits following cocaine self-administration. Brain Research. 2005 Dec 7;1064(1-2):75-82.
85. Ranaldi R, Wise RA. Blockade of D1 dopamine receptors in the ventral tegmental area decreases cocaine reward: possible role for dendritically released dopamine. Journal of Neuroscience. 2001 Aug 1;21(15):5841-6.
86. Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, et al. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012 Nov 8;491(7423):212-7.
87. Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral Tegmental Area Glutamate Neurons: Electrophysiological Properties and Projections. The Journal of Neuroscience. 2012 Oct 24;32(43):15076-85.
88. Root DH, Estrin DJ, Morales M. Aversion or salience signaling by ventral tegmental area glutamate neurons. IScience. 2018 Apr 27;2:51-62.
89. Reisi Z, Bani-Ardalan M, Zarepour L, Haghparast A. Involvement of D1/D2 dopamine receptors within the nucleus accumbens and ventral tegmental area in the development of sensitization to antinociceptive effect of morphine. Pharmacology Biochemistry and Behavior. 2014 Mar 1;118:16-21.
90. Momiyama T, Todo N, Sasa M. A mechanism underlying dopamine D1 and D2 receptor‐mediated inhibition of dopaminergic neurones in the ventral tegmental area in vitro. British Journal of Pharmacology. 1993 Aug;109(4):933-40.
91. Tanabe LM, Suto N, Creekmore E, Steinmiller CL, Vezina P. Blockade of D2 dopamine receptors in the VTA induces a long-lasting enhancement of the locomotor activating effects of amphetamine. Behavioural Pharmacology. 2004 Sep 1;15(5):387-95.
92. Ungless MA, Grace AA. Are you or aren't you? Challenges associated with physiologically identifying dopamine neurons. Trends in Neurosciences. 2012 Jul 1;35(7):422-30.
93. McGinty VB, Grace AA. Timing-dependent regulation of evoked spiking in nucleus accumbens neurons by integration of limbic and prefrontal cortical inputs. Journal of Neurophysiology. 2009 Apr;101(4):1823-35.
94. Salgado S, Kaplitt MG. The nucleus accumbens: a comprehensive review. Stereotactic and Functional Neurosurgery. 2015;93(2):75-93.
95. Zahm DS, Brog JS. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience. 1992 Oct 1;50(4):751-767.
96. Alderson HL, Parkinson JA, Robbins TW, Everitt BJ. The effects of excitotoxic lesions of the nucleus accumbens core or shell regions on intravenous heroin self-administration in rats. Psychopharmacology. 2001 Feb;153:455-63.
97. Ito R, Robbins TW, Everitt BJ. Differential control over cocaine-seeking behavior by nucleus accumbens core and shell. Nature Neuroscience. 2004 Apr 1;7(4):389-97.
98. Brog JS, Salyapongse A, Deutch AY, Zahm DS. The patterns of afferent innervation of the core and shell in the “accumbens” part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro‐gold. Journal of Comparative Neurology. 1993 Dec 8;338(2):255-78.
99. Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Annals of the New York academy of sciences. 1999 Jun;877(1):49-63.
100. Andén NE, Jackson DM. Locomotor activity stimulation in rats produced by dopamine in the nucleus accumbens: potentiation by caffeine. Journal of Pharmacy and Pharmacology. 1975 Sep;27(9):666-70.
101. Austin MC, Kalivas PW. Dopaminergic involvement in locomotion elicited from the ventral pallidum/substantia innominata. Brain Research. 1991 Feb 22;542(1):123-31.
102. Boye SM, Grant RJ, Clarke PB. Disruption of dopaminergic neurotransmission in nucleus accumbens core inhibits the locomotor stimulant effects of nicotine and D-amphetamine in rats. Neuropharmacology. 2001 May 1;40(6):792-805.
103. Cadoni C, Solinas M, Di Chiara G. Psychostimulant sensitization: differential changes in accumbal shell and core dopamine. European Journal of Pharmacology. 2000 Jan 24;388(1):69-76.
104. Kalivas PW, Weber B. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. The Journal of Pharmacology and Experimental Therapeutics. 1988 Jun;245(3):1095-102.
105. Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Research. 1990 May 14;516(1):99-106.
106. Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P. Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens. Proceedings of the National Academy of Sciences of the United States of America. 2009 Feb 24;106(8):2915-20.
107. Svenningsson P, Greengard P. A master regulator in the brain. The Scientist. 2006.
108. Todtenkopf MS, Carreiras T, Melloni Jr RH, Stellar JR. The dorsomedial shell of the nucleus accumbens facilitates cocaine-induced locomotor activity during the induction of behavioral sensitization. Behavioural Brain Research. 2002 Apr 11;131(1-2):9-16.
109. Weiner I, Gal G, Rawlins JN, Feldon J. Differential involvement of the shell and core subterritories of the nucleus accumbens in latent inhibition and amphetamine-induced activity. Behavioural Brain Research. 1996 Nov 1;81(1-2):123-33.
110. Zahm DS. Functional‐anatomical implications of the nucleus accumbens core and shell subterritories. Annals of the New York Academy of Sciences. 1999 Jun;877(1):113-128.
111. Murphy ER, Robinson ES, Theobald DE, Dalley JW, Robbins T. Contrasting effects of selective lesions of nucleus accumbens core or shell on inhibitory control and amphetamine‐induced impulsive behaviour. European Journal of Neuroscience. 2008 Jul;28(2):353-63.
112. Escobar AP, Cornejo FA, Olivares‐Costa M, González M, Fuentealba JA, Gysling K, et al. Reduced dopamine and glutamate neurotransmission in the nucleus accumbens of quinpirole‐sensitized rats hints at inhibitory D2 autoreceptor function. Journal of Neurochemistry. 2015 Sep;134(6):1081-90.
113. Pattij T, Janssen MC, Vanderschuren LJ, Schoffelmeer AN, Van Gaalen MM. Involvement of dopamine D 1 and D 2 receptors in the nucleus accumbens core and shell in inhibitory response control. Psychopharmacology. 2007 Apr;191:587-98.
114. Joyce EM, Stinus L, Iversen SD. Effect of injections of 6-OHDA into either nucleus accumbens septi or frontal cortex on spontaneous and drug-induced activity. Neuropharmacology. 1983 Sep;22(9):1141-5.
115. Kelly PH, Iversen SD. Selective 6OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. European Journal of Pharmacology. 1976 Nov;40(1):45-56.
116. Garcia‐Keller C, Martinez SA, Esparza MA, Bollati F, Kalivas PW, Cancela L. Cross‐sensitization between cocaine and acute restraint stress is associated with sensitized dopamine but not glutamate release in the nucleus accumbens. European Journal of Neuroscience. 2013 Mar;37(6):982-95.
117. Shin JH, Adrover MF, Wess J, Alvarez VA. Muscarinic regulation of dopamine and glutamate transmission in the nucleus accumbens. Proceedings of the National Academy of Sciences. 2015 Jun 30;112(26):8124-9.
118. Taber MT, Fibiger HC. Electrical stimulation of the prefrontal cortex increases dopamine release in the nucleus accumbens of the rat: modulation by metabotropic glutamate receptors. Journal of Neuroscience. 1995 May 1;15(5):3896-904.
119. Taber MT, Das S, Fibiger HC. Cortical regulation of subcortical dopamine release: mediation via the ventral tegmental area. Journal of Neurochemistry. 1995 Sep;65(3):1407-10.
120. You ZB, Tzschentke TM, Brodin E, Wise RA. Electrical stimulation of the prefrontal cortex increases cholecystokinin, glutamate, and dopamine release in the nucleus accumbens: an in vivo microdialysis study in freely moving rats. Journal of Neuroscience. 1998 Aug 15;18(16):6492-500.
121. Feja M, Hayn L, Koch M. Nucleus accumbens core and shell inactivation differentially affects impulsive behaviours in rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014 Oct 3;54:31-42.
122. West EA, Carelli RM. Nucleus accumbens core and shell differentially encode reward-associated cues after reinforcer devaluation. Journal of Neuroscience. 2016 Jan 27;36(4):1128-39.
123. Baracz SJ, Rourke PI, Pardey MC, Hunt GE, McGregor IS, Cornish JL. Oxytocin directly administered into the nucleus accumbens core or subthalamic nucleus attenuates methamphetamine-induced conditioned place preference. Behavioural Brain Research. 2012 Mar 1;228(1):185-93.
124. Jang JK, Kim WY, Cho BR, Lee JW, Kim JH. Microinjection of ghrelin in the nucleus accumbens core enhances locomotor activity induced by cocaine. Behavioural Brain Research. 2013 Jul 1;248:7-11.
125. Carpenter MB. Human neuroanatomy. 7th ed. Baltimore: The Williams and Wilkens Company; 1976.
126. White NM. Some highlights of research on the effects of caudate nucleus lesions over the past 200 years. Behavioural Brain Research. 2009 Apr 12;199(1):3-23.
127. Kennard MA. Experimental analysis of the functions of the basal ganglia in monkeys and chimpanzees. Journal of Neurophysiology. 1944 Mar 1;7(2):127-48.
128. Mettler FA, Mettler CC. The effects of striatal injury. Brain: A Journal of Neurology. 1942.
129. Divac I. Functions of the caudate nucleus. Acta Biologiae Experimentalis (Warsaw). 1968 Jan 1;28(2):107-20.
130. Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annual Review of Neuroscience. 2011 Jul 21;34:441-66.
131. Graveland GA, DiFiglia M. The frequency and distribution of medium-sized neurons with indented nuclei in the primate and rodent neostriatum. Brain Research. 1985 Feb 1;327(1-2):307-11.
132. Calabresi P, Picconi B, Tozzi A, Ghiglieri V, Di Filippo M. Direct and indirect pathways of basal ganglia: a critical reappraisal. Nature Neuroscience. 2014 Aug;17(8):1022-30.
133. Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008 Nov 26;60(4):543-54.
134. Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetamine-like psychostimulants. Brain Research Reviews. 1997 Oct 1;25(2):192-216.
135. White FJ, Hu XT, Zhang XF, Wolf ME. Repeated administration of cocaine or amphetamine alters neuronal responses to glutamate in the mesoaccumbens dopamine system. Journal of Pharmacology and Experimental Therapeutics. 1995 Apr 1;273(1):445-54.
136. Kelly PH, Seviour PW, Iversen SD. Amphetamine and apomorphine responses in the rat following 6-OHDA lesions of the nucleus accumbens septi and corpus striatum. Brain Research. 1975 Sep 5;94(3):507-22.
137. Venkataraman SS, Claussen C, Joseph M, Dafny N. Concomitant behavioral and PFC neuronal activity recorded following dose-response protocol of MPD in adult male rats. Brain Research Bulletin. 2017 Apr 1;130:125-37.
138. Cheramy A, Romo R, Godeheu G, Baruch P, Glowinski J. In vivo presynaptic control of dopamine release in the cat caudate nucleus—II. Facilitatory or inhibitory influence ofl-glutamate. Neuroscience. 1986 Dec 1;19(4):1081-90.
139. Leurquin‐Sterk G, Ceccarini J, Crunelle CL, Weerasekera A, de Laat B, Himmelreich U, et al. Cerebral dopaminergic and glutamatergic transmission relate to different subjective responses of acute alcohol intake: an in vivo multimodal imaging study. Addiction bology. 2018 May;23(3):931-44.
140. Lorenz RC, Gleich T, Buchert R, Schlagenhauf F, Kühn S, Gallinat J. Interactions between glutamate, dopamine, and the neuronal signature of response inhibition in the human striatum. Human Brain Mapping. 2015 Oct;36(10):4031-40.
141. Nair AG, Gutierrez-Arenas O, Eriksson O, Jauhiainen A, Blackwell KT, Kotaleski JH. Modeling intracellular signaling underlying striatal function in health and disease. Progress in Molecular Biology and Translational Science. 2014 Jan 1;123:277-304.
142. McGaugh JL, Herz MJ. Memory consolidation. San Francisco: Albion; 1972.
143. White NM. Effect of nigrostriatal dopamine depletion on the post-training, memory-improving action of amphetamine. Life Sciences. 1988 Jan 1;43(1):7-12.
144. McKee BL, Kelley AE, Moser HR, Andrzejewski ME. Operant learning requires NMDA-receptor activation in the anterior cingulate cortex and dorsomedial striatum, but not in the orbitofrontal cortex. Behavioral Neuroscience. 2010 Aug;124(4):500.
145. Packard MG. Glutamate infused posttraining into the hippocampus or caudate-putamen differentially strengthens place and response learning. Proceedings of the National Academy of Sciences. 1999 Oct 26;96(22):12881-6.
146. Battisti JJ, Shreffler CB, Uretsky NJ, Wallace LJ. NMDA antagonists block expression of sensitization of amphetamine-and apomorphine-induced stereotypy. Pharmacology Biochemistry and Behavior. 2000 Oct 1;67(2):241-6.
147. Cousins V, Rose JE, Levin ED. IV nicotine self-administration in rats using a consummatory operant licking response: sensitivity to serotonergic, glutaminergic and histaminergic drugs. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014 Oct 3;54:200-5.
148. Dixon ML, Thiruchselvam R, Todd R, Christoff K. Emotion and the prefrontal cortex: An integrative review. Psychological Bulletin. 2017 Oct;143(10):1033.
149. Gao M, Liu CL, Yang S, Jin GZ, Bunney BS, Shi WX. Functional coupling between the prefrontal cortex and dopamine neurons in the ventral tegmental area. Journal of Neuroscience. 2007 May 16;27(20):5414-21.
150. Imperato A, Honore T, Jensen LH. Dopamine release in the nucleus caudatus and in the nucleus accumbens is under glutamatergic control through non-NMDA receptors: a study in freely-moving rats. Brain Research. 1990 Oct 22;530(2):223-8.
151. Kalivas PW. A role for glutamate transmission in addiction to psychostimulants. Addiction Biology. 2000 Jul;5(3):325-9.
152. Kelsey JE, Willmore EJ. Electrolytic Lesions of the Nucleus Accumbens Enhance Locomotor Sensitization to Nicotine in Rats. Behavioral Neuroscience. 2006;120(3):600-11
153. Clark D, Overton PG. Alterations in excitatory amino acid-mediated regulation of midbrain dopaminergic neurones induced by chronic psychostimulant administration and stress: relevance to behavioural sensitization and drug addiction. Addict Biol. 1998 Apr;3(2):109-35.
154. Lipska BK, Al-Amin HA, Weinberger DR. Excitotoxic lesions of the rat medial prefrontal cortex: effects on abnormal behaviors associated with neonatal hippocampal damage. Neuropsychopharmacology. 1998 Dec;19(6):451-64.
155. Ramos M, Goñi-Allo B, Aguirre N. Ibotenic acid lesions of the medial prefrontal cortex block the development and expression of 3, 4-methylenedioxymethamphetamine-induced behavioral sensitization in rats. Behavioural Brain Research. 2005 May 28;160(2):304-11.
156. Wolf M, Dahlin S, Hu XT, Xue CJ, White K. Effects of lesions of prefrontal cortex, amygdala, or fornix on behavioral sensitization to amphetamine: comparison with N-methyl-D-aspartate antagonists. Neuroscience. 1995 Nov 1;69(2):417-39.
157. Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol. 1998 Apr;54(6):679-720.
158. Andrews GD, Lavin A. Methylphenidate increases cortical excitability via activation of alpha-2 noradrenergic receptors. Neuropsychopharmacology. 2006 Mar;31(3):594-601.
159. Berridge CW, Spencer RC. Differential cognitive actions of norepinephrine a2 and a1 receptor signaling in the prefrontal cortex. Brain Research. 2016 Jun 15;1641:189-96.
160. Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Molecular Neurobiology. 2002 Oct;26:203-16.
161. Dietz DM, Dietz KC, Nestler EJ, Russo SJ. Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry. 2009 May;42(S 01):S69-78.
162. Gronier B. In vivo electrophysiological effects of methylphenidate in the prefrontal cortex: involvement of dopamine D1 and alpha 2 adrenergic receptors. European Neuropsychopharmacology. 2011 Feb 1;21(2):192-204.
163. Arnsten AF. Toward a new understanding of attention-deficit hyperactivity disorder pathophysiology: an important role for prefrontal cortex dysfunction. CNS Prugs. 2009 Nov;23:33-41.
164. Seamans JK, Yang CR. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Progress in Neurobiology. 2004 Sep 1;74(1):1-58.
165. Bjijou Y, De Deurwaerdere P, Spampinato U, Stinus L, Cador M. D-amphetamine-induced behavioral sensitization: effect of lesioning dopaminergic terminals in the medial prefrontal cortex, the amygdala and the entorhinal cortex. Neuroscience. 2002 Feb 14;109(3):499-516.
166. Carter CJ, Pycock CJ. Behavioural and biochemical effects of dopamine and noradrenaline depletion within the medial prefrontal cortex of the rat. Brain Research. 1980 Jun 16;192(1):163-76.
167. Beyer CE, Steketee JD. Dopamine depletion in the medial prefrontal cortex induces sensitized-like behavioral and neurochemical responses to cocaine. Brain Research. 1999 Jul 3;833(2):133-41.
168. Bicks LK, Koike H, Akbarian S, Morishita H. Prefrontal cortex and social cognition in mouse and man. Frontiers in Psychology. 2015 Nov 26;6:1805.