Commentary
Effector CD4+ T cells (i.e. Th1, Th2, Th17) are essential in the adaptive immune system’s specific elimination of different classes of pathogens, such as viruses, bacteria, and parasites, while regulatory T cells shut these inflammatory responses off once a pathogen has been cleared [1]. Interestingly, effector T cells preferentially utilize and require aerobic glycolysis for their energetic demands, while regulatory T cells use oxidative phosphorylation [2,3]. Other immune cells follow this paradigm, including macrophage polarization between the “classical,” pro-inflammatory M1 phenotype, which utilizes aerobic glycolysis and the pentose phosphate pathway for its energetic requirements, and the “alternative,” anti-inflammatory and regenerative M2 phenotype, which relies on oxidative phosphorylation [4-6]. These studies, as well as many others, elegantly demonstrate the interdependent relationship between metabolism and immune function.
Recent studies from our laboratory and many others have demonstrated that the post-translational modification, O-GlcNAc (O-linked N-acetylglucosamine), provides another layer of metabolic regulation influencing immune cell function [7]. O-GlcNAcylation is the only known form of intracellular glycosylation. The modification dynamically cycles on and off nuclear, cytoplasmic, and mitochondrial proteins under the direction of two enzymes: O-GlcNAc transferase (OGT), which adds the modification, and O-GlcNAcase (OGA), which removes the modification [8]. OGT uses UDP-GlcNAc, the end product of the hexosamine biosynthetic pathway (HBP), to modify serine and threonine residues [9]. The HBP shunts about 2-5% of glucose from the glycolytic pathway in the making of UDP-GlcNAc. In addition, the production of UDP-GlcNAc requires input from amino acid, fatty acid, and nucleic acid metabolic pathways [10]. Because of this role in integrating various metabolic signals essential for determining T cell function, O-GlcNAc unsurprisingly is essential for proper immune cell development and function as discussed previously [11].
Further evidence of essential roles for O-GlcNAc in the normal function and development of immune cells has recently been uncovered. Building on previous work, Woo, et al. used a new, higher quality method for analyzing glycoproteins by mass spectrometry, termed isotope tagged glycoproteomics, to identify over a thousand proteins that are O-GlcNAcylated during T cell receptor activation in human cells [12]. Similar to T cell receptor signaling, B cell receptor signaling requires O-GlcNAcylation, specifically on Lyn, a critical, proximal signaling kinase [13], for full activation and proliferation of B cells [14]. OGT knock-out specifically in CD19 expressing cells prevented development of B cell precursors as well as induced apoptosis of mature and germinal center B cells, blunting antibody production [14]. Finally, a couple of groups showed that proper regulation of O-GlcNAc is essential for hematopoiesis, particularly of T cell and red blood cell precursors [15,16].
While O-GlcNAc clearly plays foundational roles in immune cell development, activation, and proliferation, we are curious if abnormal protein O-GlcNAcylation promotes or ameliorates immune cell-mediated diseases, particularly in metabolic diseases such as obesity or diabetes. If so, could O-GlcNAc sites or its two processing enzymes, OGT and OGA, potentially be manipulated therapeutically in the future? In this commentary, we will discuss recent, intriguing studies supporting both anti- and pro-inflammatory roles of O-GlcNAc in immune cell function (Figure 1) and a brief discussion of how to reconcile these differences.
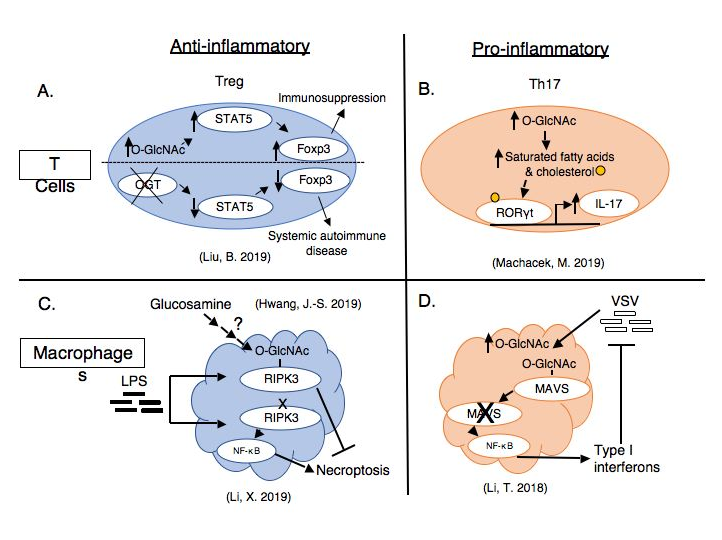
Anti-inflammatory roles of O-GlcNAc
Recent work shows O-GlcNAc has critical roles in 1) regulatory T cell stability and function, and 2) dampening macrophage responses to inflammatory stimuli.
Regulatory T cells are identified and maintained by expression of the lineage-defining transcription factor, Forkhead box P3 (Foxp3) as well as CD25, the IL-2 receptor. Foxp3 is stabilized by O-GlcNAcylation, likely by preventing ubiquitin-mediated degradation [17]. Cremediated knockout of OGT in Foxp3-expressing cells in a mouse model led to systemic autoimmune disease dominated by excessive interferon-gamma secretion [17]. Interestingly, the number of OGT-deficient regulatory T cells did not change, but rather the cells had significantly diminished signaling through the IL-2/STAT5 pathway and decreased upregulation of STAT5-regulated genes necessary for regulatory T cell immunosuppressive function [17]. While OGT deficiency’s effect on antiinflammatory cytokine secretion is not shown, decreased cellular expression of co-inhibitory cell surface markers such as PD-1 and KLRG1 contribute to the impaired immunosuppressive function in OGT-deficient cells [17]. The addition of a constitutively active STAT5 to the OGTdeficient Foxp3-expressing cells resulted in significant recovery of regulatory T cell function and attenuated the systemic autoimmune disease [17]. Using a highly specific pharmacological inhibitor of O-GlcNAcase, Thiamet-G (TMG) [18], elevated cellular O-GlcNAc levelsand increased STAT5-regulated genes in both murine and human wild-type regulatory T cells [17]. In a process distinct from intracellular O-GlcNAcylation, O-GlcNAc modification of extracellular domains of membrane proteins on regulatory T cells appears to have a similar role in promoting regulatory T cell activation through Notch signaling [19]. Knock-out of the O-GlcNAc transferase (eOGT) responsible for extracellular glycosylation exacerbated liver inflammation in a chemically-induced, autoimmune hepatitis model by impairing regulatory T cell function [19]. Thus, regulatory T cells require appropriate O-GlcNAcylation for optimal immunosuppression.
In addition to T cell responses, O-GlcNAc has critical roles in macrophages, key cells in the innate immune system. Macrophages constantly sample the environment for pathogens both by phagocytosis and subsequent digestion of proteins for antigen presentation to CD4+ T cells, as well as by responding to foreign extracellular signals, such as lipopolysaccharide (LPS) shed from the outer membrane of some bacteria [20]. Unlike activation of T and B cells [21], activation of macrophages—specifically by LPS—induces a decrease in O-GlcNAcylation, as glucose is preferentially shunted through the glycolytic pathway rather than the HBP [22]. Macrophage specific knock-out of OGT results in enhanced NF-κB activation and inflammatory cytokine secretion and worsened septic complications from intraperitoneal LPS injection or bowel puncture [22]. Additional macrophagespecific knock-out of RIPK3, an essential initiator of necroptosis, prevented the inflammatory response induced by OGT-deficient macrophages [22]. The authors identified an O-GlcNAcylation site on RIPK3, which blocked protein-protein interactions necessary for the initiation of necroptosis [22]. Mutating RIPK3’s O-GlcNAc site enhanced necroptosis and LPS-induced septic inflammation [22]. Interestingly, another group found that glucosamine treatment improved survival in the same sepsis models [23]. Glucosamine bypasses the rate-limiting step in the HBP, leading to increased UDPGlcNAc levels and protein O-GlcNAcylation [24]. These results could be explained by the now elucidated RIPK3 signaling mechanism.
Pro-inflammatory roles of O-GlcNAc
Recent work has also elucidated how O-GlcNAc can promote inflammation in both adaptive and innate immunity as well as in several different disease models.
CD4+ T helper 17 (Th17) cells normally function to eliminate fungal and persistent bacterial infections, but this strongly pro-inflammatory function also implicates Th17 cells in the pathogenesis of several autoimmune diseases, including multiple sclerosis, rheumatoid arthritis, and inflammatory bowel disease [25]. We found that a mixed population of CD4+ cells, including a mix of effector and regulatory T cells, secreted significantly more inflammatory IL-17, the eponymous Th17 cytokine, when activated ex vivo after pharmacological inhibition of OGA with TMG [7]. Because O-GlcNAc levels are sensitive to nutrient status and Th17 cells partially underlie the pathogenesis of obesity and diabetes induced chronic inflammation, we used a Western diet fed mouse model to further investigate this finding. CD4+ T cells from obese mice with elevated blood glucose had elevated O-GlcNAc levels and secreted more IL-17 than mice fed standard chow [7]. Treatment of CD4+ T cells from obese animals with TMG ex vivo further exacerbated IL-17 secretion [7]. Retinoic acid receptor-related orphan receptor gamma (RORγt), the lineage-defining transcription factor for Th17 cells, remains bound to the IL-17 gene promoter for a longer time with TMG treatment [7]. Uniquely, RORγt’s transcriptional activity can be modulated by fatty acid and cholesterol ligands, which we found were increased with TMG treatment [7]. O-GlcNAcylation of acetyl CoA carboxylase 1 (ACC1) the rate-limiting enzyme in fatty acid synthesis, may contribute to changes in the lipid microenvironment that favor RORγt activity and IL-17 secretion [7]. Thus, abnormal O-GlcNAcylation induced by systemic metabolic disease may cause abnormal cellular metabolism in Th17 cells, in this case inflammatory lipid generation, that exacerbates inflammatory cytokine secretion.
Several recent studies further support a role for O-GlcNAc in aggravating inflammation in metabolic diseases, such as obesity and diabetes. In the first, obese women with inflamed white adipose tissue were more likely to have decreased glutamine levels [26]. Supplementation with glutamine reduced the inflammatory macrophage and T cell response in the white adipose tissue by reducing metabolic flow through the hexosamine biosynthetic pathway and O-GlcNAcylation of nuclear proteins driving inflammatory genes [26]. Glutamine is a necessary substrate for the enzyme that controls the rate-limiting step in the HBP [9]; thus, the finding of decreased UDPGlcNAc and protein O-GlcNAcylation in a setting of glutamine administration is intriguing and warrants further investigation. In the second, streptozocininduced diabetes resulted in increased O-GlcNAc levels in the reproductive organs of female mice [27]. Increased inflammatory cytokine release from uterine epithelium reduced regulatory T cells migration to the uterus [27]. The diabetic mice had reduced rates of conception, and the embryos that did implant had abnormal maturation [27]. This study may provide mechanistic insight into why diabetic women have reduced ability to conceive and higher miscarriage rates and posits O-GlcNAc as a mediator of pathologic uterine inflammation. Finally, high glucose conditions increase OGT expression and protein O-GlcNAcylation in human aortic endothelial cells [28]. Down-regulation of microRNA 200a and 200b, which are known lead to degradation of OGT transcripts, were observed and mimics of these microRNAs were able to restore homeostatic levels of OGT in endothelial cells of db/db mice [28]. These studies provide continued evidence of O-GlcNAc’s pro-inflammatory effects in metabolic disease.
In addition to metabolic disease, O-GlcNAc regulates inflammatory signaling pathways in other diseases, including airway disease and breast cancer. Fibroblast growth factor 23 (FGF23) is a known pro-inflammatory mediator of diseases like chronic obstructive pulmonary disease and cystic fibrosis [29,30]. FGF23 signaling through phospholipase Cγ resulted in nuclear translocation of specific isoforms of the transcription factor, NFAT (nuclear factor of activated T cells), leading to inflammatory cytokine IL-6 secretion [29]. This group recently discovered FGF23 signaling increases O-GlcNAcylation in human bronchial epithelial cells [31]. O-GlcNAcylation of NFAT promotes its nuclear translocation and transcriptional activation of the il-6 gene [31]. Accordingly, pharmacological inhibition or siRNA targeting of OGT reduced IL-6 secretion from human bronchial epithelial cells, while OGA inhibition increased IL-6 secretion [31]. In progesterone receptor (PR)-positive breast cancer, OGT and progesterone receptor (PR) associate, leading to O-GlcNAcylation of PR [32]. Increased O-GlcNAcylation of PR results in increased transcriptional activity of PR, particularly in interferon signaling genes, whose products are known to increase resistance to chemotherapy and radiation [32-34].
Finally, O-GlcNAc has shown inflammatory functions in cells of the innate immune system, most recently macrophages and mast cells. The same group identifying an anti-inflammatory role of macrophages stimulated with LPS via O-GlcNAcylated RIPK3 discovered O-GlcNAc initiated inflammatory pathways when macrophages were virally infected [35]. In contrast to LPS, infection with vesicular stomatitis virus (VSV) led to increased HBP flux and increased protein O-GlcNAcylation within macrophages [35]. O-GlcNAcylation of MAVS, an adaptor protein important in the regulation of signaling pathways, allowed its ubiquitin-mediated degradation. Loss of MAVS led to subsequent activation of NF-κB and interferon regulatory factor (IRF) pathways, leading to increased type I interferon transcription and enhanced antiviral defense [35]. In addition to macrophages, signaling through the leukotriene 4 receptor on a human mast cell line increased O-GlcNAcylation and OGT expression and markers necessary for mast cell migration and proinflammatory IL-8 release [36]. Treatment of human mast cells with either pharmacologic or siRNA inhibition of OGT prevented inflammatory mast cell responses to leukotriene 4 [36]. Thus, spanning both adaptive and innate arms of the immune system and several disease models, O-GlcNAc functions to exacerbate inflammation.
Friend or foe: where do we go from here?
The immune system is a complex network of cells that can activate, migrate, proliferate, differentiate, and may exhibit plasticity as they encounter new environments or signals. Thus, it is often necessary to study homogenous populations of immune cells in isolation to obtain clear, reproducible results. These studies are informative and allow elucidation of important mechanisms but may lack physiological relevance by isolating cells from the many interactions that are likely to occur amongst all the varied immune and tissue cells. Thus, it is simplistic to look at whole cell or organismal alterations in O-GlcNAc as uniformly favorable or unfavorable. Untangling the incredible complexity of how multiple signals converge to alter intracellular protein O-GlcNAcylation remains a major challenge for understanding how O-GlcNAc alleviates or aggravates inflammatory responses. Despite the enormity of the task, there is good reason to be optimistic about future studies.
With the decoding of the human genome 20 years ago, science ushered in a new era of “big data.” In the span of a couple of decades, the “-omics” have become commonplace and integral scientific tools. The ability to not only obtain but develop models to integrate metabolomic, proteomic, and single cell genomic data will be critical for refining our understanding of how O-GlcNAc integrates metabolic and environmental signals to finetune immune cell function. As we continue to decode this post-translational modification’s effects, therapeutic interventions will become clearer. In T cells specifically, O-GlcNAc modifying therapies may be a feasible, not-sodistant reality. Conceivably, a similar delivery system as used in chimeric antigen receptor (CAR) T cell therapy could be used to treat immune diseases characterized by aberrant O-GlcNAcylation. A patient’s own isolated T cells could be treated with O-GlcNAc altering agents, activated, and allowed to proliferate into the desired phenotype before being re-introduced into the patient. In fact, an OGA inhibitor is already being used in phase I human clinical trials for studies in neurodegenerative disease [37]. Future studies are needed to continue untangling the interaction of metabolism, O-GlcNAcylation, and immune cell function and uncover therapeutic opportunities.
References
2. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG, Rathmell JC. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. The Journal of Immunology. 2011 Mar 15;186(6):3299-303.
3. Macintyre AN, Gerriets VA, Nichols AG, Michalek RD,Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, Rathmell JC. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell metabolism. 2014 Jul 1;20(1):61-72.
4. Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages glucose transporter 1 (GLUT1)- mediated glucose metabolism drives a proinflammatory phenotype. Journal of Biological Chemistry. 2014 Mar 14;289(11):7884-96.
5. Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, Utsumi S. Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1.Infection and immunity. 1996 Jan 1;64(1):108-12.
6. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, Pearce EJ. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015 Mar 17;42(3):419-30.
7. Machacek M, Saunders H, Zhang Z, Tan EP, Li J, Li T, Villar MT, Artigues A, Lydic T, Cork G, Slawson C. Elevated O-GlcNAcylation enhances pro-inflammatory Th17 function by altering the intracellular lipid microenvironment. Journal of Biological Chemistry. 2019 May 31;294(22):8973-90.
8. Hart GW. Nutrient regulation of signaling and transcription. Journal of Biological Chemistry. 2019 Feb 15;294(7):2211-31.
9. Hart GW, Slawson C, Ramirez-Correa G, Lagerlof O.Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry. 2011 Jul 7;80:825-58.
10. Hardivillé S, Hart GW. Nutrient regulation of signaling, transcription, and cell physiology by O-GlcNAcylation.Cell metabolism. 2014 Aug 5;20(2):208-13.
11. Machacek M, Slawson C, Fields PE. O-GlcNAc: a novel regulator of immunometabolism. Journal of bioenergetics and biomembranes. 2018 Jun 1;50(3):223-9.
12. Woo CM, Lund PJ, Huang AC, Davis MM, Bertozzi CR, Pitteri SJ. Mapping and quantification of over 2000 O-linked glycopeptides in activated human T cells with isotope-targeted glycoproteomics (Isotag). Molecular & Cellular Proteomics. 2018 Apr 1;17(4):764-75.
13. Niiro H, Clark EA. Regulation of B-cell fate by antigen-receptor signals. Nature Reviews Immunology. 2002 Dec;2(12):945-56.
14. Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung KH, Wang YH, Angata T, Lin KI. O-GlcNAcylation is required for B cell homeostasis and antibody responses. Nature communications. 2017 Nov 30;8(1):1854.
15. Abramowitz LK, Harly C, Das A, Bhandoola A, Hanover JA. Blocked O-GlcNAc cycling disrupts mouse hematopoeitic stem cell maintenance and early T cell development. Scientific reports. 2019 Aug 29;9(1):1-2.
16. Zhang Z, Parker MP, Graw S, Novikova LV, Fedosyuk H, Fontes JD, Koestler DC, Peterson KR, Slawson C. O-GlcNAc homeostasis contributes to cell fate decisions during hematopoiesis. Journal of Biological Chemistry. 2019 Jan 25;294(4):1363-79.
17. Liu B, Salgado OC, Singh S, Hippen KL, Maynard JC, Burlingame AL, Ball LE, Blazar BR, Farrar MA, Hogquist KA, Ruan HB. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nature communications. 2019 Jan 21;10(1):1-3.
18. Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nature chemical biology. 2012 Apr;8(4):393.
19. Hao X, Li Y, Wang J, Ma J, Zhao S, Ye X, He L, Yang J, Gao M, Xiao F, Wei H. Deficient O-GlcNAc glycosylation impairs regulatory T cell differentiation and notch signaling in autoimmune hepatitis. Frontiers in immunology. 2018 Oct 9;9:2089.
20. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F, Seifi B, Mohammadi A, Afshari JT, Sahebkar A. Macrophage plasticity, polarization, and function in health and disease. Journal of cellular physiology. 2018 Sep;233(9):6425-40.
21. Golks A, Tran TT, Goetschy JF, Guerini D. Requirement for O-linked N-acetylglucosaminyltransferase in lymphocytes activation. The EMBO journal. 2007 Oct 17;26(20):4368-79.
22. Li X, Gong W, Wang H, Li T, Attri KS, Lewis RE, Kalil AC, Bhinderwala F, Powers R, Yin G, Herring LE. O-GlcNAc Transferase suppresses inflammation and necroptosis by targeting receptor-interacting serine/threonine-protein kinase 3. Immunity. 2019 Mar 19;50(3):576-90.
23. Hwang JS, Kim KH, Park J, Kim SM, Cho H, Lee Y, Han IO. Glucosamine improves survival in a mouse model of sepsis and attenuates sepsis-induced lung injury and inflammation. Journal of Biological Chemistry. 2019 Jan 11;294(2):608-22.
24. Shen DL, Gloster TM, Yuzwa SA, Vocadlo DJ. Insights into O-linked N-acetylglucosamine ([0-9] O-GlcNAc) processing and dynamics through kinetic analysis of O-GlcNAc transferase and O-GlcNAcase activity on protein substrates. Journal of Biological Chemistry. 2012 May 4;287(19):15395-408.
25. Stadhouders R, Lubberts E, Hendriks RW. A cellular and molecular view of T helper 17 cell plasticity in autoimmunity. Journal of Autoimmunity. 2018 Feb 1;87:1-5.
26. Petrus P, Lecoutre S, Dollet L, Wiel C, Sulen A, Gao H, Tavira B, Laurencikiene J, Rooyackers O, Checa A,Douagi I. Glutamine Links Obesity to Inflammation in Human White Adipose Tissue. Cell Metabolism. 2020 Feb 4;31(2):375-90.
27. Brown HM, Green ES, Tan TC, Gonzalez MB, Rumbold AR, Hull ML, Norman RJ, Packer NH, Robertson SA, Thompson JG. Periconception onset diabetes is associated with embryopathy and fetal growth retardation, reproductive tract hyperglycosylation and impaired immune adaptation to pregnancy. Scientific reports. 2018 Feb 1;8(1):1-3.
28. Lo WY, Yang WK, Peng CT, Pai WY, Wang HJ. MicroRNA-200a/200b modulate high glucose-induced endothelial inflammation by targeting O-linked N-acetylglucosamine transferase expression. Frontiers in physiology. 2018 Apr 18;9:355.
29. Krick S, Grabner A, Baumlin N, Yanucil C, Helton S, Grosche A, Sailland J, Geraghty P, Viera L, Russell DW, Wells JM. Fibroblast growth factor 23 and Klotho contribute to airway inflammation. European Respiratory Journal. 2018 Jul 1;52(1):1800236.
30. Krick S, Baumlin N, Aller SP, Aguiar C, Grabner A, Sailland J, Mendes E, Schmid A, Qi L, David NV, Geraghty P. Klotho inhibits interleukin-8 secretion from cystic fibrosis airway epithelia. Scientific reports. 2017 Oct 30;7(1):1-3.
31. Krick S, Helton ES, Hutcheson SB, Blumhof S, Garth JM, Denson RS, Zaharias RS, Wickham H, Barnes JW. FGF23 Induction of O-linked N-acetylglucosamine regulates IL-6 secretion in human bronchial epithelial cells. Frontiers in endocrinology. 2018 Nov 27;9:708.
32. Trinca GM, Goodman ML, Papachristou EK, D’Santos CS, Chalise P, Madan R, Slawson C, Hagan CR. O-GlcNAc-dependent regulation of progesterone receptor function in breast cancer. Hormones and Cancer. 2018 Feb 1;9(1):12-21.
33. Weichselbaum RR, Ishwaran H, Yoon T, Nuyten DS, Baker SW, Khodarev N, Su AW, Shaikh AY, Roach P, Kreike B, Roizman B. An interferon-related gene signature for DNA damage resistance is a predictive marker for chemotherapy and radiation for breast cancer. Proceedings of the National Academy of Sciences. 2008 Nov 25;105(47):18490-5.
34. Minn AJ. Interferons and the immunogenic effects of cancer therapy. Trends in immunology. 2015 Nov 1;36(11):725-37.
35. Li T, Li X, Attri KS, Liu C, Li L, Herring LE, Asara JM, Lei YL, Singh PK, Gao C, Wen H. O-GlcNAc transferase links glucose metabolism to MAVS-mediated antiviral innate immunity. Cell host & microbe. 2018 Dec 12;24(6):791-803.
36. Min A, Lee YA, Kim KA, Shin MH. BLT1-mediated O-GlcNAcylation is required for NOX2-dependent migration, exocytotic degranulation and IL-8 release of human mast cell induced by Trichomonas vaginalissecreted LTB4. Microbes and infection. 2018 Jun 1;20(6):376-84.
37. Selnick HG, Hess JF, Tang C, Liu K, Schachter JB, Ballard JE, Marcus J, Klein DJ, Wang X, Pearson M, Savage MJ. Discovery of MK-8719, a potent O-GlcNAcase inhibitor as a potential treatment for tauopathies. Journal of medicinal chemistry. 2019 Sep 5;62(22):10062-97.