Abstract
Galectin-3 (Gal-3), the only chimera-like galectin, has three structural domains: (a) the NH2 terminal domain containing serine phosphorylation, important for nuclear localization, secretion and oligomerization; (b) a sequence susceptible to metalloprotease (MMP) cleavage; and (c) a C-terminal domain containing the carbohydrate recognition domain (CRD) and an anti-death motif. In turn, oligodendrocytes (OLG) are the resident cells responsible for CNS myelination. OLG undergo morphological and molecular changes along several maturational stages. In this context, the present review summarizes current knowledge of Gal-3 role in OLG differentiation, myelination and remyelination in experimental models of multiple sclerosis (MS). Recombinant Gal-3 (rGal-3) accelerates both OLG differentiation, evidenced by an increase in the number of mature cells to the detriment of immature ones, and actin cytoskeleton dynamics. These changes respond to rGal-3 influence on Akt, Erk 1/2, and ß-catenin signaling pathways. Our most recent results reveal a key temporal window spanning OPC and pre-OLG states for this pro-differentiating action of rGal-3 and identify several targets for rGal-3 binding including proteins related to the cytoskeleton, signaling pathways, metabolism and intracellular trafficking. Gal-3 expression in microglial cells during CPZ-induced demyelination and upon the onset of remyelination favors an M2 phenotype, hence fostering myelin debris phagocytosis through TREM-2b phagocytic receptor and MMP activity modulation, leading to OLG differentiation and remyelination. This evidence indicates that Gal-3 mediates the glial crosstalk and thus fosters remyelination both by driving OLG differentiation and promoting a phagocytic microglial phenotype. These studies also shed light on some of the mechanisms underlying Gal-3 action and open doors for the identification of new Gal-3-regulated pathways to control OLG proliferation and differentiation. Altogether, these data unveil the therapeutic potential of Gal-3 in demyelinating diseases such as MS and may allow the development of new targets.
Keywords
Galectin-3, Oligodendrocytes, Myelination, Remyelination, Microglia, Cytoskeleton, Cuprizone
Introduction
Galectins (Gals) are a group of 15 proteins characterized by a highly conserved carbohydrate-recognition domain (CRD) and made up of approximately 130 amino-acids which bind β-galactose in glycoconjugates. Gals are classified into three groups according to their structures [1-3], i.e. proto, chimera and tandem-repeat. Proto Gals, which have a single CRD, include Gal-1, Gal-2, Gal- 5, Gal-7, Gal-10, Gal-11, Gal-13, Gal-14, and Gal-15. In turn, tandem-repeat Gals contain two similar CRD and comprise Gal-4, Gal-6, Gal-8, Gal-9, and Gal-12. The only member of the chimera class, Gal-3 has three structural domains: (a) the NH2 terminal domain containing serine phosphorylation, important for nuclear localization, secretion and oligomerization; (b) a sequence susceptible to metalloprotease (MMP) cleavage; and (c) a C-terminal domain containing the CRD and an anti-death motif [4,5]. Worth pointing out, the N-terminal domain allows the formation of pentamers upon the interaction of Gal-3 monomers with glycoproteins or glycolipids.
Gal-3 has been associated to physiological processes including immunomodulation and cell proliferation, adhesion, migration, growth and differentiation [6,7], all largely determined by Gal-3 cellular localization, specific tissue, or specific pathological condition. Gal- 3 can be found in the nucleus, on the cell surface, in the extracellular space [8,9] and, depending on the cell type, also in exosomes or microvesicles [10-12] Given that Gal-3 lacks a signal sequence which may guide its translocation to the endoplasmic reticulum and further enable the classical secretory pathway, the secretion of Gal-3 proceeds in a non-classical fashion [9].
Galectin-3 in the Nervous System
Gal-3 has shown neuronal and glial expression [13]. The nerve growth factor (NGF) mediates Gal-3 expression in mouse dorsal root ganglia (DRG) neurons and macrophagelike cells, which suggests a role in the promotion of neurite outgrowth, neural cell adhesion [14,15], and neurite stabilization in the cerebellum [16]. Gal-3 is expressed in inflammatory cells, including astrocytes, microglia, macrophages, dendritic cells, eosinophils, mast cells, NK cells, and activated T and B cells. Increasing evidence has recently suggested a dual modulatory role for Gal-3 in neuroinflammation and neurodegeneration [17,18]. Gal-3 promotes microglia phagocytic capacity and chemotactic properties, thus amplifying the immune response by binding to the CCR receptor [19,20]. Upon activation, Gal-3-expressing microglia undertake myelin debris phagocytosis through FcgR, CR3/MAC-1 and SRAI/II receptors and foster remyelination upon injury or disease [21]. Gal-3 has been also recently shown to activate phagocytosis by targeting the cytoskeleton twice: first, by advancing cofilin activation, enabling filopodia and lamellipodia to extend and engulf myelin debris; and second, by advancing actin/myosin-based contraction through K-Ras.GTP/PI3K signaling, hence causing filopodia and lamellipodia to retract and internalize myelin debris [22].
Oligodendroglial biology
Oligodendrocytes (OLG) are resident central nervous system (CNS) cells in charge of myelination, i.e. the physiological process through which axons are insulated and thus provided with metabolic and trophic support for rapid saltatory conduction of action potentials [23]. Starting from a highly proliferative PDGFRα- and NG2- positive bipolar cells, oligodendroglial progenitors (OPC) later become pre-OLG, more ramified cells expressing CNPase, Olig 1 and O4, to finally develop into mature OLG (m-OLG) with MBP, APC and PLP expression and the ability to form myelin membranes [24,25].
Although Gal-3 mRNA expression has not yet been evaluated in OLG, Gal-3 has been immunocytochemically detected in rat primary cultures of OPC and m-OLG, with higher levels in the latter [26]. This Gal-3 appears to be cleaved in OPC by MMP2 but stabilized in m-OLG, which suggests variations in its biological activity during OLG differentiation [26]. Recombinant Gal-3 (rGAl-3) treatment in OPC promotes dose-dependent OLG differentiation [26], which may respond to secretion by Gal-3-expressing microglia during normal oligodendrogenesis [26,27]. In agreement, treatment with conditioned media from wild type (WT) microglia, but not Gal-3 knockout (LGALS-/-) microglia, promotes OLG differentiation. These results are tightly associated to the glycoconjugates present in OPC cell surfaces, as glycosylation signature analysis has shown that only OPC possess a permissive glycophenotype expressing the carbohydrates required for Gal-3 binding [26].
The participation of cytoskeleton dynamics in oligodendroglial differentiation and myelination has been proposed as a two-stage model of actin dynamics: first, pro-polymerizing actin cytoskeleton dynamics promote OPC branching up to full OLG maturity; second, the actin cytoskeleton shifts to depolymerizing dynamics, which triggers myelination. These stages are partly regulated by the relative levels of MBP and actin disassembly proteins colfilin-1 and gelsolin, sequestered and inactivated by phosphatidylinositol 4, 5-bisphosphate (PIP2) in the plasma membrane. MBP then competes for PIP2 binding with cofilin-1 and gelsolin and displaces them to initiate the disassembly of actin filaments in m-OLG [28]. In turn, rGal-3 has been shown to accelerate the differentiation of purified OPC, as evidenced by an early increase in m-OLG markers and a decrease in immature OLG ones [29]. Worth highlighting, these results were accompanied by the acceleration of actin dynamics in the two-stage model described above, as evidenced by an earlier polymerization peak and subsequent depolymerization. In parallel, these studies revealed an increase in Akt activation and β-catenin, MBP and gelsolin levels, together with a decrease in Erk1/2 activation [29]. Along these lines, recent reports have shown that the Akt/mTORC and Erk1/2 pathways play independent and cooperative roles in OLG differentiation along development and adulthood both in vitro and in vivo [30], and that Erk 1/2 inhibition favors OLG generation and recovery in demyelinating diseases [31].
In this context, studies using a single pulse of rGal-3 at different stages of oligodendroglial maturation were conducted to identify the temporal window of rGal-3 action and unravel its direct targets promoting differentiation. Results showed that rGal-3 promotes OLG differentiation at OPC and pre-OLG stages, generating an increase in pAkt, β-catenin, and F-actin [32]. The phosphorylation of substrate p4EB-P1 also indicated that mTORC1 is activated by rGal-3 treatment, which contributes to MBP expression and OLG maturation [33]. However, rGal- 3 treatment at the m-OLG stage failed to increase MBP expression, F-actin and β-catenin levels, or Akt activation, and produced lesser mTORC1 signaling activation. Strikingly, results also revealed an increase in gelsolin and a decrease in pErk 1/2 at all stages, both indicative of OLG differentiation [34]. Taken together, these interesting results suggest that the action of rGal-3 depends on the repertoire of glycolipids and glycoproteins present at the time of treatment.
Proteins directly interacting with rGal-3 at OPC and pre-OLG stages
Given rGal-3 pro-differentiating action at OPC and pre- OLG stages, studies were further carried out on direct rGal-3-molecule interaction at these two stages through co-immunoprecipitation and subsequent identification by mass spectrometry [32]. At the OPC stage, interacting molecules connected with cell proliferation included casein kinase II subunit alpha (CKII), proliferating cell nuclear antigen (PCNA) and voltage-dependent anionselective channel protein 1 (VDAC1), which, through rGal-3 inhibition or activation, may limit proliferation to induce OLG differentiation [35-37]. As for cytoskeletal proteins, rGal-3 was observed to interact with ras-related C3 botulinum toxin substrate 1 (Rac1) and its inhibitor, rho GDP-dissociation inhibitor 1 (RhoGDI), which may support rGal-3-dependent activation of Rac1 [29]. Most importantly, direct interaction was detected between rGal-3 and gelsolin, which is consistent with cytoplasmic gelsolin expression in OPC differentiation and maturation and suggests that cytoskeleton severing proteins may be crucial for morphological changes required for OLG differentiation [34]. In terms of energy metabolism and lipids, these assays provided evidence of rGal-3 interaction with transaldolase (TAL) and ATP-citrate synthase, which are involved in the production of the large amount of lipids necessary for myelin synthesis and protection from the deleterious effects of free radicals [38]. Regarding signaling pathways, rGal-3 was found to interact with carboxypeptidase E (CPE), which modulates the Wnt- β-catenin pathway and β-catenin levels, and with poly- ADP-ribosepolymerase 1 (PARP1), whose inhibition in the cuprizone (CPZ) model and in MS type III lesions leads to an attenuation of demyelination by increasing Akt activity [39] and favoring the oligodendroglial lineage in the subventricular zone (SVZ) [40]. Finally, on the subject of intracellular trafficking, rGal-3 interacted with Rab- 5A, which may be involved in rGal-3 internalization and redistribution in different subcellular compartments.
In contrast to the OPC stage, no proteins involved in proliferation were found to interact with rGal-3 at the pre- OLG stage. Among cytoskeletal proteins, rGal-3 was found to interact with JUP, Rac1, Arp3, gelsolin, F-actin-capping protein subunit alpha-1 (CAPZA1), actin depolymerizing protein, and septin-11 (SEPT11) and septin-7 (SEPT7), cytoskeletal GTPases promoting the formation of actin filaments. rGal-3 also interacted with microtubule-related proteins such as cytoplasmic dynein 1 light intermediate chain 2 (DYNC1LI2) and microtubule-actin cross-linking factor 1 (MACF1), with calmodulin (CAM), involved in MBP-actin interaction [41], and with stathmin (STMN1), which promotes OLG branching [33]. Taken together, these interactions emphasize the key role played by rGal- 3 in the regulation of actin filament dynamics. In turn, signaling pathways interacting with rGal-3 included nucleoside diphosphate kinase A (NME1), whose function is to inhibit OPC differentiation [42] and, strikingly, many subunits of the proteasome, which is known to degrade β-catenin, profilin 1, gelsolin, and MBP in OLG [43-45]. This suggests an inhibitory relationship between rGal-3 and these subunits of the proteasome, which may explain the increase in the levels of several proteins found upon rGal-3 treatment. Furthermore, previous studies from our laboratory indicate that inhibition of the proteasome leads to OLG differentiation and promotes remyelination in the CPZ model [46,47]. Interestingly, rGal-3 also interacted with inositol monophosphatase 1 (IMPA1), involved in the supply of inositol for phosphoinositide synthesis, which plays a crucial role in the dynamics of MBP-gelsolin- F-actin. Of note, rGal-3 was also found to interact with Erk2, which may explain the decrease in Erk activation induced by rGal-3 treatment, perhaps by blocking the phosphorylation site or generating conformational changes which prevent its activation. Interaction was also found with protein quaking, which regulates the splicing, export and stability of mRNA and protein translation, and is necessary for MBP stability and correct actin polymerization in OLG [48]. Last, regarding intracellular traffic, rGal-3 again interacted with Rab proteins. Altogether, this evidence suggests that extracellular rGal-3 is internalized and then redistributed together with target molecules to the corresponding subcellular compartments where it mediates cytoskeleton dynamics, proliferation, lipid synthesis, and signaling pathways necessary to drive OLG differentiation.
Myelination
Electron microscopy morphometric analysis have revealed a critical role for Gal-3 during myelination, reflected by LGALS3-/-mice hypomyelination, lesser myelin integrity and abnormal compaction, associated with substantial behavioral alterations [26]. To elucidate the role of Gal- 3 in myelination in vivo, studies by our group focused on Gal-3 expression at postnatal day 5 (P5), P10, P15 and P20 using transgenic mice expressing the enhanced green fluorescent protein (EGFP) driven by the promoter of oligodendroglial protein CNPase (CNP–EGFP). Gal- 3 expression showed substantial changes during white matter development, with high expression levels at P5 and a reduction along myelination. In agreement, high levels of Gal-3 were found at P5 in microglial cells localized in the corpus callosum (CC) and cingulum, fairly close to CNPase cells. Microglia appear in developing fiber tracts prior to the onset of myelination, and the transient generation of the bulk of OLG in white matter coincides with the presence of amoeboid microglia in the CC. Evidence available suggests that this ameboid microglia may have a role not only in clearing a path for axons, but also in clearing cell debris during gliogenesis. In addition, a recent study using microglia depletion has revealed an essential role for postnatal microglia in proper OPC and OLG development and homeostasis [49]. Gal-3 colocalizes with CNPase cells with mature-OLG-like morphology and has been detected at low levels in astrocytes at P10 and P15, and at high levels in the SVZ at all ages evaluated. The absence of Gal-3 affects the migration of neuroblasts from the SVZ through the rostral migratory stream toward the olfactory bulb [50] by a mechanism implying increased phosphorylation and activation of epidermal growth factor receptor (EGFR). Gal-3 also promotes the proliferation of cultured neural progenitors and its inhibition decreases the proliferative response of the SVZ after brain ischemia [51]. Our group has also proven Gal-3 to promote cell commitment toward the oligodendroglial lineage in neurosphere cultures [26]. However, Gal-3 overexpression with electroporation in the SVZ induces no inflammation in healthy postnatal gliogenesis, with a larger percentage of striatal astrocytes but a smaller percentage of OLG [52].
Gal-3 in MS and its experimental models
Oligodendroglial injury leads to demyelination, which is followed by the formation of new myelin sheaths as a regenerative response –a process referred to as remyelination [53]. This process has been described in animal models and in human demyelinating diseases such as MS [54-56]. MS disease progression varies considerably among patients, although the most frequent clinical presentation involves recurring symptoms followed by total or partial recovery, namely classic relapsing-remitting MS. Symptoms become progressive in around 50% of untreated patients and lead to clinical deterioration for several years, a stage identified as secondary progressive MS. However, about 15% of MS patients present relentless disease progression as from onset, which constitutes primary progressive MS [57-59]. The progressive stage partly responds to incomplete remyelination, which produces the loss of axonal metabolic support and concomitant axonal and neural degeneration, leading to progressive disability [60,61].
The endogenous and exogenous roles of Gals in glial cells upon demyelination/remyelination have been thoroughly reviewed recently by Jong et al. [62]. Serum from patients with secondary progressive MS has been recently shown to contain auto-antibodies against Gal-3, which may be responsible for blood brain barrier (BBB) progressive damage [63]. Actually, membrane-bound Gal-3 in human brain microvascular endothelial cells is a target for autoantibodies present in secondary progressive MS serum [64]. In addition, OPC treated with cerebrospinal fluid (CSF) from patients with primary progressive MS present a significantly more ramified morphology than control or relapsing remitting MS CSF, and a pro-differentiating transcriptome: downregulation of PDGFRα and LINGO1 genes and upregulation of MAG and, interestingly, LGALS3 genes, which establishes a link between Gal-3 upregulation and increased OPC branching. Gal-3 upregulation has also been observed in post-mortem brain tissue of patients with primary progressive MS [65]. These findings indicate that Gal-3 is a positive target for OLG differentiation in MS. However, the presence of anti-Gal-3 auto-antibodies is responsible for BBB damage, a negative effect which might be counteracted through neutralizing therapy.
Although experimental demyelination models fail to fully replicate the complexity and heterogeneity of MS, they have rendered fruitful results and allowed the development of various treatments. Several wellestablished experimental demyelination models include those mediated by immunity, viruses, and toxins. The most widely used animal model of CNS demyelination, experimental autoimmune encephalomyelitis (EAE) consists in the immunization of mice with myelin oligodendroglial glycoprotein. LGALS3-/-mice display clearly diminished CNS macrophage infiltration during EAE, which leads to lower production of pro-inflammatory cytokines interleukin IL-17 and IFN-γ and lesser disease severity [18]. Gal-3 is also involved in IL-4-mediated macrophage alternative polarization, and its effect in EAE may be thus attributed to microglial activation and proliferation [66,18]. An increase in Gal-3 expression has been reported in macrophages and microglia in the CNS of EAE mice [67], which was then reduced by copolymer 1 through an increase in antigen-specific Th2 response and secretion of IL-10, and a diminished production of proinflammatory cytokines and Gal-3.
Gal-3 is induced in several cell types involved in damaged axon and cell debris removal and axon regeneration and remyelination, which hints at a neuroprotective role of Gal-3 in EAE mice [68]. In addition, Gal-3 key role in the phagocytosis of disrupted myelin has been also reported in Wallerian degeneration, as evidenced by increased Gal-3 expression in myelin phagocytic microglia and its absence in non-myelin phagocytic ones [22,69,70]. Lack of Gal–3 actually accelerates Wallerian degeneration by modifying toll-like receptor and pro-inflammatory cytokine expression in the injured sciatic nerve [71].
MS-induced inflammation may decrease SVZ cell proliferation and thus hinder repair. Gal-3 expression increases in active human MS lesions [72], in periventricular regions in human MS and after a virus-induced MS model, in which the loss of Gal-3 restores SVZ proliferation through a reduction in the number of immune cells [73].
Another useful demyelination model, CPZ administration produces massive demyelination by mature oligodendroglial apoptosis through pathogenic T cell-independent mechanisms. This model keeps the BBB intact [74,75] and is characterized by astroglial activation and resident microglial recruitment, whereas peripheral macrophage infiltration remains controversial [76-81]. Phagocytosis of myelin debris by microglia in CPZ demyelination, once again key to the onset of remyelination [82], is concomitant with an increase in phagocytic receptor TREM-2b expression and CD200R and TNF-α production [83].
Studies by our group using the CPZ model to determine Gal-3 participation in demyelination/remyelination [84] rendered comparable courses of demyelination in both LGALS3-/- and WT mice up to the 5th week of treatment. However, while WT mice initiated spontaneous remyelination in the 5th week of CPZ treatment, when the CPZ diet was still in place, LGALS3-/- mice underwent constant demyelination up to the 6th week, with pronounced astroglial activation. Studies of Gal-3 expression in WT mice showed its upregulation in microglia but not in astrocytes. Interestingly, only WT mice displayed activated microglia with ED1 (CD68) expression and TREM-2b upregulation during CPZ-induced demyelination, while CPZ-treated LGALS-/- mice exhibited more numerous microglia with activated caspase-3, which suggests that the absence of Gal-3 alters the microglial response against demyelination. M2-cell-conditioned medium has been shown to enhance OLG differentiation in vitro, and M2 cell depletion has been proven to impair OLG differentiation in vivo, which indicates that M2 cell polarization is a key factor for efficient remyelination [85]. Therefore, microglial Gal-3 expression may hence favor the onset of remyelination, either by inducing an M2 phenotype or exerting a direct effect on OLG differentiation.
Gal-3 and MMP modulation in remyelination
MMP-2 and MMP-9 catalyze the cleavage of Gal-3 into a 22 kDa fragment which has a CRD and a 9 kDa polypeptide containing the N-terminal domain [86,87]. This process changes the properties of the 22 kDa fragment relative to the function and binding properties of the CRD [87]. Although the relationship between Gal-3 and MMP has been mostly studied in connection with tumorigenesis and metastasis [88], MMP-9 deletion in nervous tissues has been shown to protect against ischemic brain injury, diminishing neuroinflammation and preserving BBB integrity [89]. In turn, LGALS-/- mice submitted to hypoxic brain injury show lower expression of MMP-9, while WT mice co-express Gal-3 and MMP-9 in activated microglia [90]. MMP can also induce myelin protein degradation in vitro [91-93], and are involved in postnatal myelination, myelin maintenance, and remyelination [93,94,95]. Also, MMP-3 can mediate mature OLG apoptosis and microglial activation, with the production of microglial inflammatory cytokines and the consequent exacerbation of neural cell degeneration [96]. Interestingly, WT mice show an increase in MMP-3 expression and a decrease in CD45, TNFα, and TREM-2b+ cells during remyelination, while LGALS-/-mice exhibit no changes either in demyelination or remyelination [97].
Conclusion
Gal-3 is essential for microglial polarization following CNS injury, although with different effects on different injuries probably responding to time- and contextdependent factors [98]. Some possible molecular mechanisms underlying Gal-3 neuroprotection from microglia are particularly interesting in OLG biology, myelination and remyelination processes. Oligomerized Gal-3 molecules may crosslink to IGFR upon binding to their glycans on the surface and delay their removal by endocytosis, which results in prolonged microglial mitogenic signaling [99,100]. This is particularly relevant because IGF-1 acts directly on OLG differentiation and myelination [101] and, when secreted by microglia, reduces OLG apoptosis in the CPZ model [102]. Moreover, Gal-3 involvement in alternative microglial polarization induced by IL-4, as that taking place in macrophages [103], may also explain the effect of Gal-3 on cell commitment toward the oligodendroglial lineage, as IL-4-activated microglia favor oligodendrogenesis through a mechanism mediated by IGF-I, while IFN-gamma-activated microglia favor neurogenesis [104]. Taken together, our results have shown that Gal-3 expression in microglial cells during CPZ-induced demyelination and upon the onset of remyelination favors an M2 phenotype, hence fostering myelin debris phagocytosis through TREM-2b phagocytic receptor and MMP activity modulation, leading to OLG differentiation and remyelination [84,97] (Figure 1). Given that unsuccessful remyelination may result from inefficient removal of myelin debris by microglia, unveiling the mechanisms controlling phagocytosis may prove an effective approach to reversing disability and curbing disease progression. In addition, Gal-3 has been recently identified as a novel endogenous TREM-2b ligand [105]. Most interestingly, Gal-3 has been lately shown to activate microglial phagocytosis through a mechanism involving cytoskeleton modulation [22]. In line with these studies and emphasizing the key role played by Gal-3 in the regulation of actin filament dynamics, our most recently published results showed that rGal-3 accelerates oligodendroglial differentiation also through cytoskeleton modulation [29], which was strongly supported by the identification of Gal- 3 direct interaction with a key protein in actin dynamics control such as gelsolin [32] (Figure 1). Given that Gal-3 binds to multiple targets, its effects on OLG biology and myelination may reflect direct and indirect actions, i.e. those mediated by microglia, astrocytes or peripheral cells. Nowadays, numerous reports describe the high heterogeneity and developmental and region-specific differences among microglia, astrocytes and OPC in the healthy and pathological CNS [106-108], which lead to different effects exerted by Gal-3 in these cells. Not only microglia seem to be involved in myelin phagocytosis; myelin uptake is an early response of astrocytes in diseases with prominent myelin injury, which results in the recruitment of immune cells [109]. Surprisingly, specific oligodendroglial lineage populations have been recently identified in the EAE model expressing genes involved in antigen processing and presentation (MHC-I and -II). It has also been demonstrated that OPC have phagocytic capacity and that MHC-II expressing OPC can activate memory and effector CD4+ T cells [110]. Therefore, future experiments will be highly relevant in clarifying the possible involvement of Gal-3 in phagocytic functions in astrocytes and the oligodendroglial lineage.
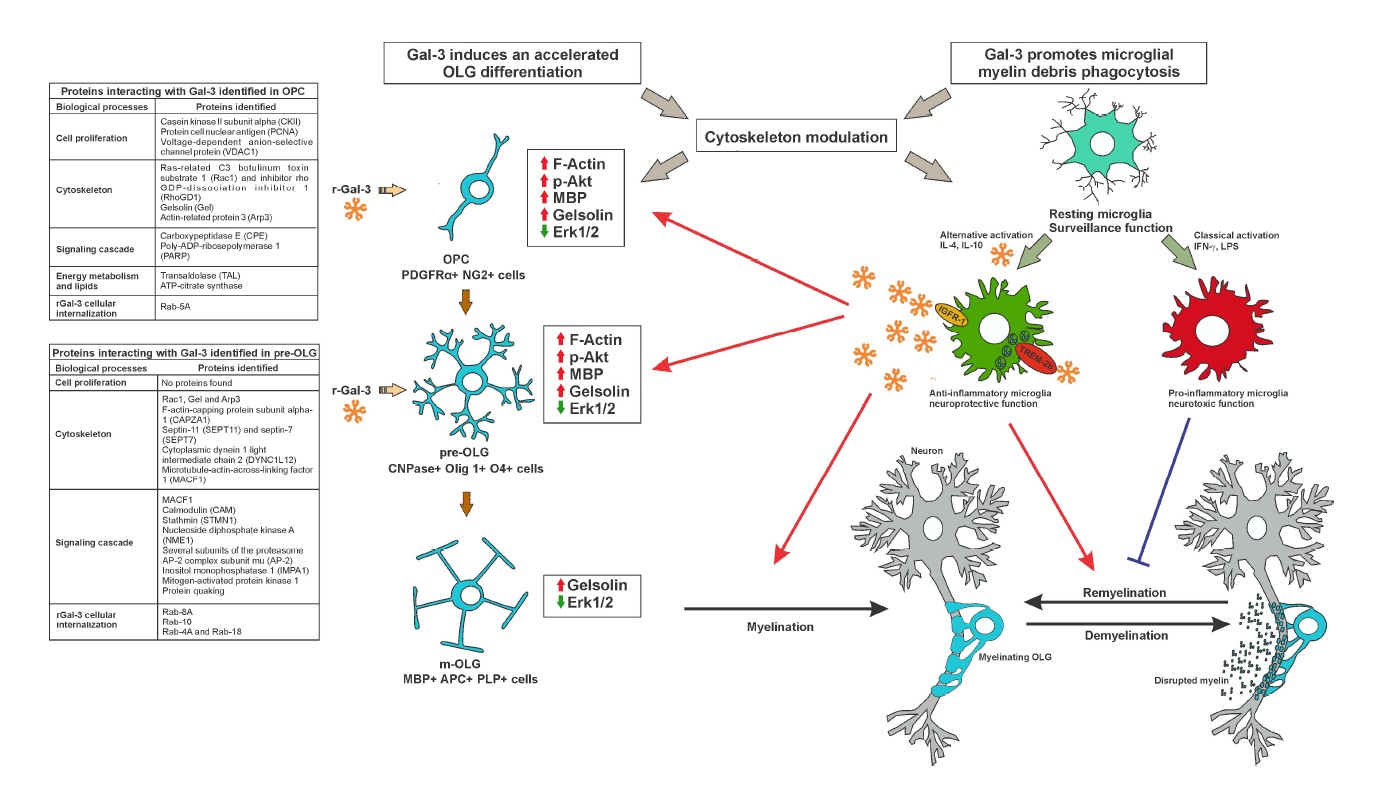
The vast evidence available so far indicates that Gal- 3 mediates the glial crosstalk and favors remyelination both by driving OLG differentiation and promoting a phagocytic microglial phenotype. These studies also shed light on some of the mechanisms underlying Gal-3 action and open doors for the identification of new signaling pathways regulated by Gal-3 to control OLG proliferation and differentiation (Figure 1). In this context, Gal-3 may constitute a therapeutic alternative, probably through the delivery of rGal-3 to OPC and microglia by nanocarriers and exosomes, as they can deliver cargo to other cells, easily cross the BBB and exhibit low immunogenicity. This is also supported by fact that Gal-3 can be excreted through exosomes. Altogether, these data unveil the therapeutic potential of Gal-3 in demyelinating diseases such as MS and may allow the development of new targets.
Author Contribution
LAP supported and wrote the manuscript.
Conflict of Interest
The author declares no conflict of interest.
Acknowledgements
The author wishes to thank Hernan Hoyos and Laura Thomas for dedicating their PhD studies to elucidating Gal-3 involvement in oligodendroglial biology. In addition, the author wants to thank Maria Marta Rancez for her help with article edition. This study was supported by grants from the Argentine Agency for the Promotion of Science and Technology (PICT 2016-0319) and the University of Buenos Aires (UBACYT- 20020170100285BA) to LAP.
References
2. Hirabayashi J, Kasai K. The family of metazoan metal– independent beta–galactoside–binding lectins: Structure, function and molecular evolution. Glycobiology. 1993 Aug;3(4):297-304. doi: 10.1093/glycob/3.4.297.
3. Yang RY, Rabinovich GA, Liu FT. Galectins: Structure, function and therapeutic potential. Expert Rev Mol Med. 2008 Jun 13;10:e17.
4. Dumic J, Dabelic S, Flögel M. Galectin-3: an openended story. Biochim Biophys Acta. 2006; 1760: 616–635.
5. Funasaka T, Raz A, Nangia-Makker P. Nuclear transport of galectin-3 and its therapeutic implications. Semin Cancer Biol. 2014; 27: 30–38.
6. Rabinovich GA, Croci DO. Regulatory circuits mediated by lectin-glycan interactions in autoimmunity and cancer. Immunity. 2012 Mar 23;36(3):322-35.
7. Di Lella S, Sundblad V, Cerliani JP, Guardia CM, Estrin DA, Vasta GR, et al. When galectins recognize glycans: from biochemistry to physiology and back again. Biochemistry. 2011 Sep 20;50(37):7842-57.
8. Wang JL, Gray RM, Haudek KC, Patterson RJ. Nucleocytoplasmic lectins. Biochim Biophys Acta. 2004 Jul 6;1673(1-2):75-93.
9. Hughes RC. Secretion of the galectin family of mammalian carbohydrate–binding proteins. Biochim Biophys Acta. 1999 Dec 6;1473(1):172-85.
10. Théry C, Boussac M, Véron P, Ricciardi–Castagnoli P, Raposo G, Garin J, et al. Proteomic analysis of dendritic cell–derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J Immunol. 2001 Jun 15;166(12):7309-18.
11. Mehul B, Hughes RC. Plasma membrane targetting, vesicular budding and release of galectin 3 from the cytoplasm of mammalian cells during secretion. J Cell Sci. 1997 May;110 ( Pt 10):1169-78.
12. Welton JL, Khanna S, Giles PJ, Brennan P, Brewis IA, Staffurth J, et al. Proteomics analysis of bladder cancer exosomes. Mol Cell Proteomics. 2010 Jun;9(6):1324-38.
13. Yoo HI, Kim EG, Lee EJ, Hong SY, Yoon CS, Hong MJ, et al. Neuroanatomical distribution of galectin–3 in the adult rat brain. J Mol Histol. 2017 Apr;48(2):133-146.
14. Pesheva P, Kuklinski S, Biersack HJ, Probstmeier R. Nerve growth factor–mediated expression of galectin–3 in mouse dorsal root ganglion neurons. Neurosci Lett. 2000 Oct 20;293(1):37-40.
15. Pesheva P, Kuklinski S, Schmitz B, Probstmeier R. Galectin-3 promotes neural cell adhesion and neurite growth. J Neurosci Res. 1998 Dec 1;54(5):639-54.
16. Mahoney SA, Wilkinson M, Smith S, Haynes LW. Stabilization of neurites in cerebellar granule cells by transglutaminase activity: identification of midkine and galectin-3 as substrates. Neuroscience. 2000;101(1):141- 55.
17. Shin T. The pleiotropic effects of galectin–3 in neuroinflammation: a review. Acta Histochem. 2013 Jun;115(5):407-11.
18. Jiang HR, Al Rasebi Z, Mensah–Brown E, Shahin A, Xu D, Goodyear CS, et al. Galectin-3 deficiency reduces the severity of experimental autoinmune encephalomyelitis. J Immunol. 2009 Jan 15;182(2):1167-73.
19. Burguillos MA, Svensson M, Schulte T, Boza-Serrano A, Garcia-Quintanilla A, Kavanagh E, et al. Microgliasecreted galectin-3 acts as a toll-like receptor 4 ligand and contributes to microglial activation. Cell Rep. 2015 Mar 10;10(9):1626-1638.
20. Wesley UV, Vemuganti R, Ayvaci ER, Dempsey RJ. Galectin-3 enhanced angiogenic and migratory potential of microglia cells via modulation of integrin linked kinase signaling. Brain Res. 2013 Feb 16;1496:1-9.
21. Rotshenker S, Reichert F, Gitik M, Haklai R, Elad- Sfadia G, Kloog Y. Galectin-3/MAC-2, Ras and PI3K activate complement receptor-3 and scavenger receptor- AI/II mediated myelin phagocytosis in microglia. Glia. 2008; 56:1607–1613.
22. Reichert F, Rotshenker S. Galectin-3 (MAC-2) Controls Microglia Phenotype Whether Amoeboid and Phagocytic or Branched and Non-phagocytic by Regulating the Cytoskeleton. Front Cell Neurosci. 2019 Mar 14;13:90.
23. Bercury KK, Macklin WB. Dynamics and mechanisms of CNS myelination. Dev Cell. 2015 Feb 23;32(4):447-58.
24. Nave KA, Werner HB. Myelination of the nervous system: mechanisms and functions. Annu Rev Cell Dev Biol. 2014;30:503-33.
25. Snaidero N, Simons M. The logistics of myelin biogenesis in the central nervous system. Glia. 2017 Jul;65(7):1021-1031.
26. Pasquini LA, Millet V, Hoyos HC, Giannoni JP, Croci DO, Marder M, et al. Galectin-3 drives oligodendrocyte differentiation to control myelin integrity and function. Cell Death Differ. 2011 Nov;18(11):1746-56.
27. Ellison J A, de Vellis J. Amoeboid microglia expressing GD3 ganglioside are concentrated in regions of oligodendrogenesis during development of the rat corpus callosum. Glia. 1995 Jun;14(2):123-32.
28. Zuchero JB, Fu MM, Sloan SA, Ibrahim A, Olson A, Zaremba A, Dugas JC, Wienbar S, Caprariello AV, Kantor C, Leonoudakis D, Lariosa-Willingham K, Kronenberg G, Gertz K, Soderling SH, Miller RH, Barres BA. CNS myelin wrapping is driven by actin disassembly. Dev Cell. 2015 Jul 27;34(2):152-67.
29. Thomas L, Pasquini LA. ExtracellularGalectin-3 Induces accelerated oligodendroglial differentiation through changes in signaling pathways and cytoskeleton dynamics. Mol Neurobiol. 2019 Jan;56(1):336-349.
30. Ishii A, Furusho M, Macklin W, Bansal R. Independent and cooperative roles of the Mek/ERK1/2-MAPK and PI3K/Akt/mTOR pathways during developmental myelination and in adulthood. Glia. 2019 Jul;67(7):1277- 1295.
31. Suo N, Guo YE, He B, Gu H, Xie X. Inhibition of MAPK/ERK pathway promotes ligodendrocytes generation and recovery of demyelinating diseases. Glia. 2019 Jul;67(7):1320-1332. doi: 10.1002/glia.23606.
32. Thomas L, Pasquini LA. Galectin-3 exerts a prodifferentiating and pro-myelinating effect within a temporal window spanning precursors and pre-oligodendrocytes: insights into the mechanisms of action. Mol Neurobiol. 2020 Feb;57(2):976-987.
33. Figlia G, Gerber D, Suter U. Myelination and mTOR. Glia. 2018 Apr;66(4):693-707.
34. Shao Z, Lee X, Huang G, Sheng G, Henderson CE, Louvard D, et al. LINGO-1 regulates oligodendrocyte differentiation through the cytoplasmic gelsolin signaling pathway. J Neurosci. 2017 Mar 22;37(12):3127-3137.
35. Zhou J, Tien AC, Alberta JA, Ficarro SB, Griveau A, Sun Y, et al. A sequentially priming phosphorylation cascade activates the gliomagenic transcription factor Olig2. Cell Rep. 2017 Mar 28;18(13):3167-3177.
36. Bastian C, Quinn J, Tripathi A, Aquila D, McCray A, Dutta R, et al. CK2 inhibition confers functional protection to young and aging axons against ischemia by differentially regulating the CDK5 and AKT signaling pathways. Neurobiol Dis. 2019 Jun;126:47-61.
37. Imada S, Yamamoto M, Tanaka K, Seiwa C, Watanabe K, Kamei Y, et al. Hypothermia-induced increase of oligodendrocyte precursor cells: Possible involvement of plasmalemmal voltage-dependent anion channel 1. J Neurosci Res. 2010 Dec;88(16):3457-66.
38. Banki K, Colombo E, Sia F, Halladay D, Mattson DH, Tatum AH, et al. Oligodendrocyte-specific expression and autoantigenicity of transaldolase in multiple sclerosis. J Exp Med. 1994 Nov 1;180(5):1649-63.
39. Veto S, Acs P, Bauer J, Lassmann H, Berente Z, Setalo G Jr, Borgulya G, et al. Inhibiting poly(ADP-ribose) polymerase: a potential therapy against oligodendrocyte death. Brain. 2010 Mar;133(Pt 3):822-34.
40. Plane JM, Grossenbacher SK, Deng W. PARP-1 deletion promotes subventricular zone neural stem cells toward a glial fate. J Neurosci Res. 2012 Aug;90(8):1489- 506.
41. Boggs JM, Rangaraj G. Interaction of lipid-bound myelin basic protein with actin filaments and calmodulin. Biochemistry. 2000 Jul 4;39(26):7799-806.
42. Owlanj H, Jie Yang H,Wei Feng Z. Nucleoside diphosphate kinase Nm23-M1 involves in oligodendroglial versus neuronal cell fate decision in vitro. Differentiation. 2012 Nov;84(4):281-93.
43. Choi YN, Lee SK, Seo TW, Lee JS, Yoo SJ. C-terminus of Hsc70-interacting protein regulates profilin1 and breast cancer cell migration. Biochem Biophys Res Commun. 2014 Apr 18;446(4):1060-6.
44. Ni XG, Zhou L, Wang GQ, Liu SM, Bai XF, Liu F, et al. The ubiquitin-proteasome pathway mediates gelsolin protein downregulation in pancreatic cancer. Mol Med. Sep-Oct 2008;14(9-10):582-9.
45. Belogurov A Jr, Kudriaeva A, Kuzina E, Smirnov I, Bobik T, Ponomarenko N, et al. Multiple sclerosis autoantigen myelin basic protein escapes control by ubiquitination during proteasomal degradation. J Biol Chem. 2014 Jun 20;289(25):17758-66.
46. Millet V, Moiola CP, Pasquini JM, Soto EF, Pasquini LA. Partial inhibition of proteasome activity enhances remyelination after cuprizone-induced demyelination. Exp Neurol. 2009 Jun;217(2):282-96.
47. Pasquini LA, Paez PM, Moreno MA, Pasquini JM, Soto EF. Inhibition of the proteasome by lactacystin enhances oligodendroglial cell differentiation. J Neurosci. 2003 Jun 1;23(11):4635-44.
48. Doukhanine E, Gavino C, Haines JD, Almazan G, Richard S. The QKI-6 RNA binding protein regulates actin-interacting protein-1 mRNA stability during oligodendrocyte differentiation. Mol Biol Cell. 2010 Sep 1;21(17):3029-40.
49. Hagemeyer N, Hanft K, Akriditou M, Unger N, Park E, Stanley E, et al. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance During Adulthood. Acta Neuropathol. 2017 Sep;134(3):441-458.
50. Comte I, Kim Y, Young CC, van der Harg JM, Hockberger P, Bolam PJ, et al. Galectin-3 maintains cell motility from the subventricular zone to the olfactory bulb. J Cell Sci. 2011 Jul 15;124(Pt 14):2438-47.
51. Yan YP, Lang BT, Vemuganti R, Dempsey RJ. Galectin-3 mediates post-ischemic tissue remodeling. Brain Res. 2009 Sep 8;1288:116-24.
52. Al-Dalahmah O, Campos Soares L, Nicholson J, Draijer S, Mundim M, Lu VM, et al. Galectin-3 modulates postnatal subventricular zone gliogenesis. Glia. 2020 Feb;68(2):435-450.
53. Franklin RJM, Ffrench-Constant C. Regenerating CNS myelin from mechanisms to experimental medicines. Nat Rev Neurosci. 2017 Nov 16;18(12):753-769.
54. Prineas JW, Barnard RO, Kwon EE, Sharer LR, Cho ES. Multiple sclerosis: remyelination of nascent lesions. Ann Neurol. 1993 Feb;33(2):137-51.
55. Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006 Dec;129(Pt 12):3165-72.
56. Patani R, Balaratnam M, Vora A, Reynolds R. Remyelination can be extensive in multiple sclerosis despite a long disease course. Neuropathol Appl Neurobiol. 2007 Jun;33(3):277-87.
57. Bjartmar C, Kinkel PR, Kidd G, Rudick RA, Trapp BD. Axonal loss in normal-appearing white matter in a patient with acute MS. Neurology. 2001 Oct 9;57(7):1248-52.
58. McFarland HF, Martin R. Multiple sclerosis: A complicated picture of autoimmunity. Nat Immunol. 2007 Sep;8(9):913-9.
59. Tallantyre EC, Bo L, Al-Rawashdeh O, Owens T, Polman CH, Lowe JS, et al. Clinico pathological evidence that axonal loss underlies disability in progressive multiple sclerosis. Mult Scler. 2010 Apr;16(4):406-11.
60. Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010 Apr;11(4):275-83.
61. Franklin RJM, ffrench-Constant C, Edgar JM, Smith KJ. Neuroprotection and repair in multiple sclerosis. Nat Rev Neurol. 2012 Nov 5;8(11):624-34.
62. de Jong CGHM, Gabius HJ, Baron W. The emerging role of galectins in (re)myelination and its potential for developing new approaches to treat multiple sclerosis. Cell Mol Life Sci. 2020 Apr;77(7):1289-1317.
63. Nishihara H, Shimizu F, Kitagawa T, Yamanaka N, Akada J, Kuramitsu Y, et al. Identification of galectin-3 as a possible antibody target for secondary progressive multiple sclerosis. Mult Scler. 2017 Mar;23(3):382-394.
64. Dietrich JB. The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. J Neuroimmunol. 2002 Jul;128(1-2):58-68.
65. Haines JD, Vidaurre OG, Zhang F, Riffo-Campos ÁL, Castillo J, Casanova B, et al. Multiple sclerosis patient-derived CSF induces transcriptional changes in proliferating oligodendrocyte progenitors. Mult Scler. 2015 Nov;21(13):1655-69.
66. Radosavljevic G, Volarevic V, Jovanovic I, Milovanovic M, Pejnovic N, Arsenijevic N, et al. The roles of Galectin-3 in autoimmunity and tumor progression. Immunol. Res. Apr 2012, 52(1-2):100-10.
67. Reicher F, Rotshenker S. Galectin-3/MAC–2 in experimental allergic encephalomyelitis. Exp Neurol. 1999 Dec;160(2):508-14.
68. Itabashi T, Arima Y, Kamimura D, Higuchi K, Bando Y, Takahashi-Iwanaga H, Murakami M, Watanabe M, Iwanaga T , Nio-Kobayashi J. Cell- and stage-specific localization of galectin-3, a b-galactoside-binding lectin, in a mouse model of experimental autoinmune encephalomyelitis. Neurochem Int. 2018 Sep;118:176-184.
69. Rotshenker S. The role of Galectin-3/MAC–2 in the activation of the innate–immune function of phagocytosis in microglia in injury and disease. J Mol Neurosci. 2009 Sep;39(1-2):99-103.
70. Rotshenker S. Wallerian degeneration: The innate–immune response to traumatic nerve injury. J Neuroinflammation. 2011 Aug 30;8:109.
71. Mietto BS, Jurgensen S, Alves L, Pecli C, Narciso MS, Assunção-Miranda I, et al. Lack of galectin–3 speeds Wallerian degeneration by altering TLR and pro– inflammatory cytokine expressions in injured sciatic nerve. Eur J Neurosci. 2013 May;37(10):1682-90.
72. Stancic M, van Horssen J, Thijssen VL, Gabius HJ, van der Valk P, Hoekstra D, et al. Increased expression of distinct galectins in multiple sclerosis lesions. Neuropathol Appl Neurobiol. 2011 Oct;37(6):654-71.
73. James RE, Hillis J, Adorján I, Gration B, Mundim MV, Iqbal AJ, et al. Loss of galectin-3 decreases the number of immune cells in the subventricular zone and restores proliferation in a viral model of multiple sclerosis. Glia. 2016 Jan;64(1):105-21.
74. Bakker DA, Ludwin SK. Blood-brain barrier permeability during Cuprizone-induced demyelination. Implications for the pathogenesis of immune-mediated demyelinating diseases. J Neurol Sci. 1987 Apr;78(2):125- 37.
75. Kondo A, Nakano T, Suzuki K. Blood-brain barrier permeability to horseradish peroxidase in twitcher and cuprizone-intoxicated mice. Brain Res. 1987 Nov 3;425(1):186-90.
76. Suzuki K, Kikkawa Y. Status spongiosus of CNS and hepatic changes induced by cuprizone (biscyclohexanone oxalyldihydrazone). Am J Pathol. 1969 Feb;54(2):307-25.
77. Blakemore WF. Demyelination of the superior cerebellar peduncle in the mouse induced by cuprizone. J Neurol Sci. 1973 Sep;20(1):63-72.
78. Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001 Jan;11(1):107-16.
79. McMahon EJ, Suzuki K, Matsushima GK. Peripheral macrophage recruitment in cuprizone-induced CNS demyelination despite an intact bloodbrain barrier. J Neuroimmunol. 2002 Sep;130(1-2):32-45.
80. Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch U-K, Mack M, et al. Microglia in the adult brain arise from Ly-6ChiCCR2C monocytes only under defined host conditions. Nat Neurosci. 2007 Dec;10(12):1544-53.
81. Lampron A, Larochelle A, Laflamme N, Préfontaine P, Plante MM, Sánchez MG, et al. Inefficient clearance of myelin debris by microglia impairs remyelinating processes. J Exp Med. 2015 Apr 6;212(4):481-95.
82. Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006 Jan 4;26(1):328-32.
83. Voß EV, Škuljec J, Gudi V, Skripuletz T, Pul R, Trebst C, Stangel M. Characterisation of microglia during deand remyelination: can they create a repair promoting environment? Neurobiol Dis. 2012 Jan;45(1):519-28.
84. Hoyos HC, Rinaldi M, Mendez-Huergo SP, Marder M, Rabinovich GA, Pasquini JM, Pasquini LA. Galectin-3 controls the response of microglial cells to limit cuprizoneinduced demyelination. Neurobiol Dis. 2014;62:441-455.
85. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadra van Wijngaarden P, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013 Sep;16(9):1211- 1218.
86. Ochieng J, Fridman R, Nangia-Makker P, Kleiner DE, Liotta LA, Stetler-Stevenson WG, et al. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and -9. Biochemistry. 1994 Nov 29;33(47):14109-14.
87. Ochieng J, Green B, Evans S, James O, Warfield P. Modulation of the biological functions of galectin-3 by matrix metalloproteinases. Biochim Biophys Acta. 1998 Jan 8;1379(1):97-106.
88. Takenaka Y, Fukumori T, Raz A. Galectin-3 and metastasis. Glycoconj J. 2002;19(7-9):543-549. doi:10.1023/B:GLYC.0000014084.01324.15.
89. Svedin P, Hagberg H, Sävman K, Zhu C, Mallard C. Matrix metalloproteinase-9 gene knock-out protects the immature brain after cerebral hypoxia-ischemia. J Neurosci. 2007 Feb 14;27(7):1511-8.
90. Doverhag C, Hedtjärn M, Poirier F, Mallard C, Hagberg H, Karlsson A, et al. Galectin-3 contributes to neonatal hypoxic-ischemic brain injury. Neurobiol Dis. 2010 Apr;38(1):36-46.
91. Chandler S, Cossins J, Lury J, Wells G. Macrophage metalloelastase degrades matrix and myelin proteins and processes a tumour necrosis factor-alpha fusion protein. Biochem Biophys Res Commun. 1996 Nov 12;228(2):421- 9.
92. Shiryaev SA, Savinov AY, Cieplak P, Ratnikov BI, Motamedchaboki K, Smith JW, et al. Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in autoimmune multiple sclerosis. PLoS One. 2009;4(3):e4952.
93. Hansmann F, Herder V, Kalkuhl A, Haist V, Zhang N, Schaudien D, et al. Matrix metalloproteinase-12 deficiency ameliorates the clinical course and demyelination in Theiler’s murine encephalomyelitis. Acta Neuropathol. 2012 Jul;124(1):127-42.
94. Ulrich R, Baumgärtner W, Gerhauser I, Seeliger F, Haist V, Deschl U, et al. MMP-12, MMP-3, and TIMP-1 are markedly upregulated in chronic demyelinating theiler murine encephalomyelitis. J Neuropathol Exp Neurol. 2006 Aug;65(8):783-93.
95. Skuljec J, Gudi V, Ulrich R, Frichert K, Yildiz O, Pul R, et al. Matrix metalloproteinases and their tissue inhibitors in cuprizone-induced demyelination and remyelination of brain white and gray matter. J Neuropathol Exp Neurol. 2011 Sep;70(9):758-69.
96. Kim YS, Joh TH. Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson’s disease. Exp Mol Med. 2006 Aug 31;38(4):333-47.
97. Hoyos HC, Marder M, Ulrich R, Gudi V, Stangel M, Rabinovich GA, et al. The Role of Galectin-3: From Oligodendroglial Differentiation and Myelination to Demyelination and Remyelination Processes in a Cuprizone-Induced Demyelination Model. Adv Exp Med Biol. 2016;949:311-332.
98. Rahimian R, Béland LC, Kriz J. Galectin-3: mediator of microglia responses in injured brain. Drug Discov Today. 2018 Feb;23(2):375-381.
99. Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, et al. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004 Oct 1;306(5693):120-4.
100. Lalancette-Hébert M, Swarup V, Beaulieu JM, Bohacek I, Abdelhamid E, Weng YC, et al. Galectin-3 is required for resident microglia activation and proliferation in response to ischemic injury. J Neurosci. 2012 Jul 25;32(30):10383-95.
101. Zeger M, Popken G, Zhang J, Xuan S, Lu QR, Schwab MH, et al. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia. 2007 Mar;55(4):400-11.
102. Mason JL, Jones JJ, Taniike M, Morell P, Suzuki K, Matsushima GK. Mature oligodendrocyte apoptosis precedes IGF-1 production and oligodendrocyte progenitor accumulation and differentiation during demyelination/ remyelination. J Neurosci Res. 2000 Aug 1;61(3):251-62.
103. Mackinnon AC, Gibbons MA, Farnworth SL, Leffler H, Nilsson UJ, Delaine T, et al. Regulation of transforming growth factor-ß1-driven lung fibrosis by galectin-3. Am J Respir Crit Care Med. 2012 Mar 1;185(5):537-46.
104. Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006 Jan;31(1):149-60.
105. Boza-Serrano A, Ruiz R, Sanchez-Varo R, García- Revilla J, Yang Y, Jimenez-Ferrer I, et al. Galectin-3, a novel endogenous TREM2 ligand, detrimentally regulates inflammatory response in Alzheimer’s disease. Acta Neuropathol. 2019 Aug;138(2):251-273.
106. Priller J, Prinz M. Targeting microglia in brain disorders. Science. 2019 Jul 5;365(6448):32-33.
107. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017 Jan;541(7638):481-7.
108. Foerster S, Hill M, Franklin R. Diversity in the Oligodendrocyte Lineage: Plasticity or Heterogeneity? Glia. 2019 Oct;67(10):1797-1805.
109. Ponath G, Ramanan S, Mubarak M, Housley W, Lee S, Rezan Sahinkaya F, et al. Myelin Phagocytosis by Astrocytes After Myelin Damage Promotes Lesion Pathology. Brain. 2017 Feb;140(2):399-413.
110. Falcão AM, van Bruggen D, Marques S, Meijer M, Jäkel S, Agirre E, et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat Med. 2018 Dec;24(12):1837-1844.