Abstract
Autoantibodies are antibodies produced by B cells capable of attacking their own tissues or organs after loss of immune tolerance. A large body of literature shows that autoantibodies play an important role in neurological diseases associated with immune abnormalities. Nowadays, neurological disorders are an important cause of disability and death in populations around the world, so it is important to study the role and mechanisms of autoantibodies in neurological disorders associated with immune abnormalities. The blood-brain barrier is biologically important for maintaining the normal physiological state of the central nervous system, and we have previously found that monoclonal autoantibodies of SLE origin can inhibit the expression of tight junction proteins in the blood-brain barrier. Our findings provide clues for the study of the role of autoantibodies in neurological disorders and greatly attract us to conduct a more in-depth and systematic review and synthesis. In this paper, we summarize the functions of autoantibodies in autoimmune and non-autoimmune diseases and focus on the roles and mechanisms of autoantibodies in immune-associated neurological disorders by taking central and peripheral neurological diseases as the entry points, which can provide new molecular targets and new theoretical basis for the specific diagnosis and targeted therapy of neurological diseases.
Keywords
Autoantibodies, Dysimmunity related neurological diseases, Diagnosis, Therapy, Pathogenesis
Background
The presence of the blood-brain barrier (BBB) makes it possible for the neurological effects of autoantibodies to be overlooked. Over the past decade, it has been gradually recognized that autoantibodies may play a role in the pathogenesis of neurological disorders, leading to neurological dysfunction [1]. It has been found that in a variety of neurological disorders, such as antiphospholipid antibody syndrome and neuropsychiatric lupus, the integrity and permeability of the BBB is disrupted, and the ensuing autoantibodies cross the blood-brain barrier, which in turn disrupts the neurological system and triggers cognitive dysfunction, encephalitis, and other symptoms [2,3]. Thus, autoantibodies play an important role in the pathogenesis of neurological diseases, especially autoimmune neurological diseases [4]. In neuromyelitis optica spectrum disorder (NMOSD), for example, APQ4-IgG binds to APQ4 receptors on astrocytes to undergo a chain reaction after complement activation, leading to astrocyte damage in NMOSD, which in turn disrupts the blood-brain barrier, and even leads to neuronal death [5].
However, the specific mechanism of action of autoantibodies in neuropsychiatric disorders associated with immune abnormalities, especially autoantibody-involved such as neuropsychiatric lupus, multiple sclerosis, and myasthenia gravis, as well as in numerous neurodegenerative disorders, is unclear, and therapeutic targets have not yet been defined [6]. Therefore, in-depth exploration of the mechanisms of autoantibodies in these neurological diseases and the development of related therapeutic strategies have become urgent scientific challenges. Taking this as a starting point, this review aims to summarize and deeply explore the role and mechanism of autoantibodies in immune abnormality-associated neurological diseases, to provide important clues for unraveling the pathogenesis of autoimmune neuropsychiatric disorders and to provide potential therapeutic targets for the treatment of neurological disorders, so as to fill in the gaps of the current research on autoantibodies in neurological disorders' pathogenesis.
The Discovery of Autoantibodies and Their Role in Autoimmune and Non-Autoimmune Diseases
Autoantibodies are potentially harmful antibodies that, after the body loses the integrity of its immune tolerance, become unable to recognize "self" or "non-self" antigens and attack the host by interacting with their own antigens, triggering inflammation of tissues [7,8]. The history of autoantibodies can be traced back many years to 1900, when Paul Ehrlich first proposed "the concept of horror autotoxicus", in which he pointed out that the immune system can only produce antibodies against non-self antigens, i.e. the "side-chain theory" [9]. The more widely accepted theory, however, is the doctrine of clonal selection, proposed in 1959 by the Australian immunologist Frank Macfarlane Burnet, which has been crucial to the development of highly diverse antibody libraries. This theory suggests that autoimmune diseases may be mediated by pathogenic autoantibodies due to "clonal deletion", and that pathogenic autoantibodies play a crucial role in the development of autoimmune diseases [10].
Autoantibodies not only play an important role in the pathologic process of autoimmune diseases, but also assume the role of biomarkers, which provide useful clinical information for the diagnosis and treatment of diseases [11]. In SLE, autoantibodies such as anti-dsDNA antibodies and anti-Sm antibodies are involved in the formation of immune complexes and inflammatory damage in several end organs such as kidney, skin, and central nervous system (CNS) [12]. Anti-dsDNA autoantibodies have potential for drug development against intracellular targets, and it has been suggested that their targets may be utilized in the treatment of autoimmune diseases [13]. However, the diagnostic sensitivity of anti-dsDNA antibodies and anti-Sm antibodies for SLE was 33% and 27%, respectively, and the diagnostic specificity was low, which is not only detrimental to the accurate diagnosis of autoimmune diseases, including SLE, but also fails to achieve differential diagnosis between multiple autoimmune diseases. Therefore, whether anti-dsDNA antibodies and anti-Sm antibodies and their targets are suitable for the development of targeted drugs for autoimmune diseases, we believe that the relevant studies still have certain controversies and challenges.
In rheumatoid arthritis (RA), anticitrullinated protein antibodies (ACPA) are considered to be the most diagnostic biomarker, are present in 60-70% of RA patients, and are associated with cachexia and poor prognosis of joint damage [14,15]. ACPA has a dual impact in the course of RA, not only stimulating osteoclast differentiation and triggering bone erosion, but also potentially playing an important role in long-term inflammation [16,17]. Furthermore, in IgA nephropathy, the galactose-deficient IgA1 molecule (gd-IgA1) originating from mucosal tissues leads to the formation of autoantibodies, such as anti-gd-IgA1 IgG. gd-IgA1 and gd-IgA1 IgG form immune complexes that are deposited on the glomerular thylakoid membranes, triggering the activation of a cascade of complement and cytokines, which are involved in IgA nephropathy pathogenesis. The main reason for this is that glucocorticoids (a form of immunotherapy) are not used in IgA nephropathy. Currently glucocorticoids (an immunosuppressive agent) are considered a relatively appropriate regimen for the treatment of IgA nephropathy [18,19]. In systemic sclerosis (SSc), antinuclear antibodies (ANA) are considered to be the most accurate independent predictor, present in more than 90% of SSc patients, with high diagnostic specificity and predictive value [20]. However, whether ANA is involved in the pathogenesis of SSc is unclear, ANA contains different subtypes, and the therapeutic effects of different drugs can vary when different ANA subtypes are used as therapeutic targets: if therapeutic drugs are correctly paired with the ANA subtypes, they may have an important impact on the therapeutic effects of SSc [21,22]. For example, anti-RNA polymerase III antibodies, an important subtype in ANA their possible pathogenic mechanism is to be translocated into cells to interact directly with intracellular components and receptors, and anti-RNA polymerase III antibodies as a biomarker predict autoimmune syndromes such as rapid skin thickness growth and gastric sinus vasodilatation [23]. Therefore, we hypothesized that pathogenic autoantibodies of a biomarker could serve as potential targets for SSc. By exploring the binding sites of pathogenic autoantibodies and receptors in SSc, we can precisely inhibit the binding of autoantibodies to receptors and thus block the pathogenesis of SSc. Currently, the pathogenesis of SSc involves at least 240 pathways and a large number of dysregulated proteins, so it is still very challenging to realize precision targeting therapy through this drug and ANA subtype-based pairing strategy.
Autoantibodies are widely present in a variety of pathological conditions and play a key role not only in the pathogenesis of autoimmune diseases, but also in non-autoimmune diseases such as cardiovascular and infectious diseases [24,25]. Antiphospholipid autoantibodies are a class of antibodies that specifically bind anionic phospholipids and phospholipid protein complexes. Studies have shown that in cardiovascular diseases, elevated levels of antiphospholipid autoantibodies in patients mediate monocyte adhesion, platelet aggregation, complement activation, and endothelial damage, thus playing an important role in the pathogenesis of cardiovascular diseases [26]. In addition, autoantibodies against glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) are capable of causing celiac disease [27]. Autoantibodies to GPIHBP1 block the ability of GPIHBP1 to bind and transport lipoprotein lipase, thereby interfering with lipoprotein lipase-mediated processing of triglyceride-rich lipoproteins, leading to severe celiac disease [28]. In COVID-19 and influenza patients, the presence of large amounts of autoantibodies correlates with the severity of the disease [29]. This suggests that autoantibodies may be involved in the pathophysiologic course of diseases associated with viral infections; however, the specific role of autoantibodies in the pathogenesis of these diseases is not fully understood.
Autoantibodies are Involved in the Pathogenesis of Neurological Disorders
Neurological diseases are important causes of disability and death in populations worldwide [30]. In recent years, the global mortality rate of neurological diseases has risen by nearly 30%, and their impact on human health has become more and more serious [31]. Considering that autoantibodies play a great role in both the pathogenesis and the treatment of immune abnormality-associated neurological disorders, it is particularly important to understand the roles and mechanisms of autoantibodies in neurological disorders. In this section, we have divided neurologic diseases into central nervous system and peripheral nervous system diseases, so that we can more clearly understand the role of autoantibodies in different neurologic-related diseases.
Autoantibodies and central nervous system disorders
CNS autoimmune diseases are often closely associated with target antigens on the neuronal or glial surface as well as their specific autoantibodies, which target neuronal or glial proteins on the cell surface. For example, in neuromyelitis optica system disorder (NMOSD), autoantibodies target surface water channel proteins of neuroglial cells, which in turn lead to disease. Therefore, exploring potentially pathogenic autoantibodies will help diagnose and treat patients with CNS disorders, and the following section describes the roles and mechanisms of autoantibodies in CNS disorders by detailing several CNS disorders that will offer potential tools for diagnosing and targeted treatment of CNS diseases to provide potential targets and research directions.
Neuromyelitis Spectrum disorder (NMOSD): Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune disease with symptoms such as sensory loss, visual impairment, etc. AQP4 is the astrocyte water channel protein aquaporin 4, and its autoantibody, AQP4-IgG, plays an important role in the pathogenesis of NMOSD. In patients, helper T cells together with IL-6 promote B cell differentiation and produce autoantibody AQP4-IgG [32]. When AQP4-IgG enters CNS tissues, it first binds to the AQP4 receptor on the surface of astrocytes and mediates the complement reaction leading to astrocyte damage, which then leads to granulocyte infiltration, oligodendrocyte death, and ultimately neuronal cell death, causing sensory loss and visual impairment [5].
Neuromyelitis optica (NMO) is a typical case in NMOSD. Glucose-regulated protein 78 (GRP78) is an important molecular chaperone located on the endoplasmic reticulum that is expressed on BMECs (brain microvascular endothelial cells), and non-AQP4-specific rAb IgG is a recombinant antibody in NMO patients. Binding of non-AQP4-specific rAb IgG to its target antigen as GRP78 activates BMECs through the typical NF-κB signaling pathway, resulting in decreased expression of the tight junction protein Claudin-5 and increased BBB permeability, thus facilitating the entry of the autoantibody AQP4-IgG into the brain tissue. C1q has been shown to induce neurodegenerative disease axonal damage, which leads to a decrease in neurological function, which in turn leads to a loss of immunoreactivity in spinal cord neurons [33-35]. Following the binding of AQP4-IgG to AQP4, astrocytes transcribe and secrete the complement component C3, and the content of C3 transcripts is upregulated, which increases C1q synthesis in microglia upon binding to C3a and C3b receptors on microglial cells, ultimately causing motor deficits in the early stages of NMO [36,37].
From the above, it is clear that C3 and its transcripts, especially C3a and C3b, play a key role in the mechanistic pathway of NMO occurrence in the connection between astrocytes and microglia. We hypothesized that the pathway of NMO genesis could be blocked by inhibiting the production of C3a and C3b on astrocytes in response to AQP4-IgG and AQP4. It has been shown that T cell-expressed CTSL (histatinase L) is able to process C3 into biologically active C3a and C3b, and that CTSL-specific chemical inhibitors and CTSL function-blocking antibodies inhibit CTSL-mediated C3a and C3b production [38]. Therefore, CTSL may be a potential target for the treatment of NMO, and the effect of this chemical inhibitor and antibody on C3 processing, whether it has adverse effects, and whether there are superior function-blocking substances may be a focus of research for the treatment of NMO.
Anti-NMDAR encephalitis: Anti-N-methyl-d-aspartate receptor (NMDAR) encephalitis is a synaptic autoimmune disease in which patients may develop cognitive deficits, seizures, and abnormal movements [39,40]. In this case, autoantibodies target NMDAR receptors on the synaptic surface, which in turn destroys the receptors, affecting signaling between neurons as well as the body's learning and memory functions [41-43]. In recent years, it has been shown that binding of autoantibodies against the NR1 subunit of NMDAR to NMDAR induces binding, capping, and cross-linking of NMDAR and autoantibodies from the synaptic and extra synaptic gaps, leading to internalization of NMDAR as a means of decreasing the synaptic surface NMDAR density, and that other synaptic proteins, synaptic densities, or neurotransmitter receptors are unaffected [44]. Thus, it is clear that anti-NMDAR autoantibodies are involved in anti-NMDAR encephalitis by eliminating MNDAR-mediated synaptic currents by decreasing NMDAR function but not affecting the number of synapses. In addition to this, autoantigen-mediated aberrant activation and differentiation of B cells plays a positive feedback amplifying effect in the involvement of autoantibodies in the development of anti-NMDAR encephalitis. GluN1, an antigenic peptide against NMDAR encephalitis, is involved in the development of cognitive deficits and neuropsychiatric symptoms in patients. It has been found that the GluN1 subunit of NMDAR abnormally activates B cells to produce and secrete autoantibodies into the serum, and subsequently these autoantibodies enter the brain, leading to a decrease in the selectivity and reversibility of the synaptic localization of NMDAR, resulting in cognitive deficits and neuropsychiatric symptoms in patients [44,45].
From the above, it is clear that B cells activated by the GluN1 subunit play an important role in the mechanism of anti-NMDAR encephalitis. We hypothesized that the pathway could be impeded by reducing the number of activated B cells to inhibit autoantibody production. In a recent study, NMDAR-CAAR (NMDAR-specific chimeric autoantibody receptor) T cells were shown to deplete anti-NMDAR B-cell lines and persistently reduce autoantibody levels by recognizing large numbers of human patient-derived autoantibodies, releasing effector molecules, and proliferating and selectively killing antigen-specific target cell lines [46]. Therefore, we speculate that anti-NMDAR encephalitis can be alleviated to some extent by NMDAR-CAAR therapy and serve as a future direction for precision-targeted therapy.
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS): The exact pathogenesis of ME/CFS, a complex neuroimmune disorder in which patients develop symptoms of extreme fatigue after activity, which worsens with physical or mental activity but does not fully improve with rest, is unclear. Relevant studies have shown that autoantibodies play an important role in the development of ME/CFS [47]. Both ß2AdR and M3 acetylcholine receptors are important vasodilators [48]. There is evidence that natural autoantibodies against ß2AdR and M3 acetylcholine receptors are present in about one-third of ME/CFS patients [49,50]. Specific autoantibodies in patients bind to the receptors, leading to receptor dysfunction, which in turn leads to key symptoms of ME/CFS, such as fatigue and muscle pain. In addition, anti-G protein-coupled receptor autoantibodies also play a role in ME/CFS, where anti-G protein-coupled receptor autoantibodies bind to the G protein-coupled receptor, which in turn activates the complement response and mediates inflammation, which exacerbates the symptoms of ME/CFS [48].
Currently, immunoadsorption (IA) is a relatively effective treatment when treating patients with ME/CFS. Some studies have demonstrated that clinical symptoms can be improved in some patients after cyclic IA treatment, however, its efficacy is not absolute, and some patients have little response or even deteriorate during treatment [51]. Therefore, for the time being, IA therapy is still only a clinical treatment direction and is still in the research stage. It should be noted, however, that the current study data only involves the treatment results of a small number of patients. In order to make the conclusions more scientific, the study can be further strengthened by increasing the number of reports on the outcome of patients treated with IA.
Tumors of the nervous system: Glioma is a tumor originating from glial cells in the brain. Wei et al. found that anti-Glial Fibrillary Acidic Protein (GFAP) autoantibodies can specifically target gliomas and correlate with WHO classification and tumor volume. Moreover, GFAP autoantibodies can be used as an early diagnostic marker for glioma, which has potential clinical utility [52]. Fine filament protein C (FLNC) is usually present in muscle tissues involved in the maintenance of myonodal stability. Some studies have found that FLNC is expressed in gliomas and also has a role in tumor development, and its expression level increases with tumor progression. Anti-FLNC autoantibodies were found in the serum of glioma patients, and their levels were negatively correlated with tissue expression, so serum anti-FLNC autoantibodies can also be used as a potential serum biomarker for early diagnosis of low-grade gliomas. Glutamate decarboxylase (GAD) is an intracellular enzyme expressed in neurons, and its physiological function is to decarboxylate glutamate to γ-aminobutyric acid (GABA), which is the main inhibitory neurotransmitter within the central nervous system, and when GAD is inhibited, it can increase excitability in the nervous system [53,54]. Some cases have shown that the risk of cancer in patients is increased by the presence of GAD-Abs on the cell surface of neurons, and it is suggested that patients with high levels of GAD-Abs expression should be screened for potential cancers, but whether it is involved in tumor development is not known [55,56].
In addition to anti-GFAP autoantibodies and anti-FLNC autoantibodies, classical anti-Hu, Ma2, CV2, and amphipathic protein antibodies are biomarkers for the presence of neural-related tumors. These findings suggest that autoantibodies are closely associated with neural-related tumors and have application value as tumor diagnostic biomarkers, but the specific role and mechanism of autoantibodies in tumorigenesis are still unclear, and therefore the study of autoantibodies in the treatment of tumors is still in the gap stage [57].
Anti-IgLON5 diseases: Anti-IgLON5 disease is a rare autoimmune neurological disorder whose main features include the presence of anti-IgLON5 autoantibodies and the specific accumulation of tau protein in neurons [58]. IgLON5 is a cell adhesion molecule that is widely expressed on the surface of neurons and is involved in signaling, cell adhesion, neuronal development, and synaptogenesis [59]. In vitro studies have confirmed that IgLON5 IgG may disrupt the cytoskeleton of rat hippocampal neurons, causing crosstalk between factors external to the cell and the neuronal cytoskeleton, which ultimately leads to an abnormal accumulation of neurofilaments, which leads to neuronal atrophy and axonal swelling, and thus induces neurodegenerative lesions [58]. Considering the close association between the cytoskeleton and tau proteins, it was further demonstrated that anti-IgLON5 antibody induced differentiation of human neural stem cells could cause accumulation of p-tau, increase the proportion of p-tau-positive neurons, and at the same time, disrupt the synaptic structure and function, which is involved in the development of anti-IgLON5 diseases [60].
While the exact pathogenesis of anti-IgLON5 disease is not known, and therefore no definitive treatment can be used to accurately treat the disease, it is still possible to use targeted medications to alleviate the patient's symptoms based on clinical signs. For example, the number of B-cells producing anti-IgLON5 antibodies has been found to be significantly increased in the cerebrospinal fluid of patients, so the use of anti-B-cell drugs (e.g., Rituximab) to inhibit the proliferation of B-cells may have a certain degree of efficacy [61]. Immunotherapy is a common clinical treatment for autoimmune diseases, but there are different discussions for anti-IgLON5 diseases. It has been suggested that the pathogenic mechanism of anti-IgLON5 antibodies is the destruction of the outside of the cell in relation to the cytoskeleton, rather than targeting proteins on the surface of neurons [61]. Therefore, immunotherapy may not be efficacious for patients. However, clinical data show that 41% of patients benefit from short-term immunotherapy and 75% from long-term immunotherapy [59]. Therefore, until the exact mechanism of anti-IgLON5 antibody pathogenicity is determined, immunotherapy is not an ineffective treatment.
Limbic encephalitis (LE): Limbic encephalitis (LE) caused by LGI1-IgG is an anti-neuronal surface antigen-associated autoimmune encephalitis characterized by facial and arm dystonia and epilepsy, but the associated pathogenic mechanisms mediated by autoantibodies are unknown [62,63]. LGI1 is a neuronal secretory protein that is concentrated in the hippocampus and contains an N-terminal LRR structural domain and a C-terminal EPTP structural domain. LGI1 is a neuronal secretory protein concentrated in the hippocampus and contains an N-terminal LRR structural domain and a C-terminal EPTP structural domain. It was found that LGI1 IgG reacts strongly with the LRR and EPTP structural domains of LGI1 and prevents LGI1 from interacting with ADAM22 and ADAM23, thus disrupting both pre- and postsynaptic signaling. In turn, these two receptors form a presynaptic membrane complex containing Kv1.1 channels and a postsynaptic membrane complex containing AMPAR, respectively, with LGI1. Blockade of LGI1 IgG leads to a decrease in Kv1.1 and AMPAR in the hippocampus, which in turn leads to neuronal hyperexcitability, decreased plasticity, and reversible memory deficits, yet how these proteins are down-regulated when LGI1 is dysfunctional remains unclear (PMM) [64-66].
It is clear that autoantibody disruption of LGI1-ADAM22/23 trans-synaptic complex synapses is an important part of the pathogenesis of limbic encephalitis (LE), where the regulation of the kv1.1 channel and AMPA-R is one of the important molecular mechanisms that may be involved in other neurological diseases. Exploring the pathogenesis of limbic encephalitis (LE) requires us to understand the potential role of LGI1 in the extrasynaptic context and its association with kv1.1 channels and AMPA-R, especially how LGI1 targets and down-regulates the expression of kv1.1 channels and related proteins in association with the LGI1-ADAM22/23 complex. Currently, corticosteroids are commonly used to treat anti-LGI1 encephalitis and show a favorable prognosis, notably, short-term memory deficits are present in approximately one-third of patients, suggesting that there is still a need to identify alternative treatment options to optimize recovery in the clinical setting [67].
Autoantibodies and peripheral nervous system diseases
Peripheral nervous system autoimmune diseases are often associated with demyelination problems. Myelin is a membrane that wraps around the axon of a nerve cell and consists of Schwann cells and the myelin membrane. Studies have shown that in the peripheral nervous system, myelin, when phagocytosed by macrophages, will be involved in the mechanisms of demyelinating diseases, including Guillain-Barré syndrome (GBS), chronic inflammatory demyelinating polyneuropathy (CIDP) and multiple sclerosis (MS) diseases. Currently, the mechanism of how macrophages cause demyelination is unclear, but we review and summarize the role and mechanism of autoantibodies in peripheral nerve diseases from the perspective of the role of autoantibodies and myelin, which play an important role in the development of demyelinating diseases as well as their sub-branches and provide potential targets and research directions for the diagnosis and treatment of peripheral neurological diseases [79,80].
Guillain-Barré syndrome: Guillain-Barre syndrome (GBS) is an autoimmune disease of peripheral nerves that occurs after infection and is pathologically characterized by demyelinating lesions of peripheral nerves and nerve roots and inflammatory cell infiltration of small vessels. Studies have shown that after infection by pathogens (e.g., Campylobacter jejuni), antibodies generated by immunoreactivity targeting lipid oligosaccharides on the outer membrane of Clostridium jejuni can cross-react with gangliosides GM1 and GD1a on the nerve cell membranes, which can lead to nerve damage or functional blockade of nerve conduction [61,81]. van den Berg et al. summarized the results of multiple experiments and concluded that in the pathogenesis of acute motor axonal neuropathy (AMAN), a common subtype of GBS, anti-GM1 and anti-GD1a antibodies bind to antigens located in the vicinity of the Rumphrey's junction and activate complement, followed by the formation of membrane attack complexes (MACs) that disrupt voltage-gated sodium channels. This damage can lead to detachment of the paranodal myelin sheath and subsequent failure of nerve conduction. Furthermore, they hypothesized the pathogenesis of another common subtype of GBS, acute inflammatory demyelinating polyneuropathy (AIDP), in which multiple antibodies target antigens located on the myelin sheaths, and the activation of complement by the antibodies leads to the formation of MACs on the outer surface of Schwann's cells, as well as macrophage invasion of the myelin sheaths leading to the onset of demyelinating injury [82].
Plasma exchange and intravenous immunoglobulin (IVIg) are commonly used in the treatment of GBS, but 20% of patients are left with persistent and significant disability after treatment, therefore, we still need further research to provide more appropriate treatment for patients [83]. We can learn that complement activation has a crucial role in the pathogenesis of GBS, therefore, the development of suitable complement inhibitors for the treatment of patients will likely be a new direction of treatment. New studies have found that eculizumab is a monoclonal antibody targeting complement protein C5, and in combination with IVIg, eculizumab treatment presents a safe and well-tolerated prognosis, however, the sample size of patients tested in the recent study was too small to rule out the patient variability or other clinical factors, so further clinical testing and evidence are needed for treatment with eculizumab [84-86].
Chronic Inflammatory Demyelinating Polyneuropathy: Chronic inflammatory demyelinating polyneuropathy (CIDP) is a chronic inflammatory neuropathy. Neural Fascicle Protein 155 (NF155) is a cell adhesion molecule expressed on the cell surface and is involved in the formation of septum-like connections in myelinated axonal paranodal nodes [87,88]. In CIDP, anti-Nerve Fascicle Protein 155 (NF155) antibodies are key pathogenic autoantibodies. It has been shown that anti-NF155 antibodies directly target NF155 on Schwann cells, inducing selective depletion of NF155 in peripheral nerves, which in turn prevents the normal formation of parajunctional axial glial junctions, interfering with neurotransmission, and participating in CIDP [87,89]. Abnormalities of NF155 are associated with ataxia, tremor, and poor response to intravenous immunoglobulin in patients with CIDP associated with CIDP and is often used as a biomarker for diagnosis and guidance of treatment.
Therefore, based on the newly discovered mechanism of action of anti-NF155 antibodies in CIDP, we believe that blocking the targeting of anti-NF155 antibodies to NF155 on Schwann cells or reducing the production of such antibodies may be able to achieve therapeutic goals. However, how anti-NF155 antibodies target Schwann cells and the associated complement mechanisms have not been elucidated, and studies have shown that overexpression of NF155 leads to myelin repair in a rat model established by introducing an NF155 overexpressing lentiviral vector plasmid, and the same effect was found in patients [88,90]. From this, we speculated that it might be possible to repair myelin and alleviate CIDP by up-regulating NF155 expression and thereby achieving the goal of repairing myelin. Therefore, before the discovery of the specific pathogenic mechanism of anti-NF155 antibody, the use of drugs to promote NF155 expression might be a feasible therapeutic approach.
Indeed, although some studies have pointed to autoantibodies to NF155 as an important factor in the induction of CIDP, the production of anti-NF155 antibodies has been found in only some patients with CIDP in the clinic, and the typical features of CIDP (macrophage-induced demyelination) have not been [91]. Therefore, the current therapeutic approach is also not directed against Contactin-1, but rather by subcutaneous or intravenous immunoglobulin (IVIg) [92]. Therefore, we believe that anti-NF155 antibody can be used as a biomarker for CIDP, but more research is needed to determine how to achieve therapeutic goals through anti-NF155 antibody.
Fisher syndrome: Fisher syndrome (MFS), also known as extraocular muscle paralysis-ataxia-deep reflex loss syndrome, is characterized by external ocular paresis and ataxia of the cerebellum. The disease can be caused by infection with microorganisms bearing the GQ1b epitope, such as Haemophilus influenzae. When such microorganisms enter the interior of the body, the patient's body produces corresponding autoantibodies-immunoglobulin G (IgG) and anti-GQ1b antibodies [93]. GQ1b is highly expressed in the parasympathetic ganglia and neuromuscular junctions of the oculomotor, cochlear, and abducens nerves, where anti-GQ1b antibodies bind specifically to the antigen, which activates complement, leading to paralysis of the muscles or impaired neurotransmission at this site, and ultimately to symptoms such as nystagmus, cerebellar ataxia, and so on [94,95]. Thus, Fisher syndrome is induced when IgG and anti-GQ1b antibodies bind to GQ1b antigen's expressed on the associated cranial nerves and muscle spindles.
There are many similarities between many diseases and MFS in terms of clinical presentation, and therefore many difficulties remain in clinical diagnosis and more accurate diagnostic criteria are needed for diagnosis. In 60% of MFS patients, cerebrospinal fluid protein shows a tendency to be elevated, and this feature is used to diagnose the disease, but the detection of anti-GQ1B antibodies would have more diagnostic value than the cerebrospinal fluid protein level. The existence of pathogenicity of anti-GQ1b antibodies has also given us the idea of treating the disease, and we believe that, by blocking the binding of anti-GQ1b antibodies to the GQ1b antigens on neuromuscular, we may be able to have a therapeutic effect on the disease. Studies have shown that in an in vitro mouse model of NMJ, the use of human intravenous immunoglobulin (IVIg) inhibits the binding of anti-GQ1b antibodies to GQ1b, thereby preventing the binding of autoantibodies to GQ1b at nerves, complement activation, and subsequent pathophysiological effects [96,97]. This NMJ model also provides a suitable system for studying the therapeutic effects of IVIg in antibody-mediated neuromuscular diseases. Furthermore, in MFS, the complement activation-dependent neuroexocytosis of anti-GQ1b antibody at the mouse neuromuscular junction results from massive uncontrolled calcium inward currents and is accompanied by calpain-mediated morphological disruption of nerve endings. It has been shown that both calcium depletion and calpain inhibition protect the cytoskeleton from degradation by immunohistochemical and ultrastructural analysis. Therefore, calcineurin inhibitors may be effective in limiting nerve ending damage and calcium inward flow, a key pathogenic pathway, and may be able to play a role in the treatment of autoimmune diseases of the peripheral nervous system [98,99].
Summary and Outlook
Tables 1 and 2 summarize the role of autoantibodies in the pathogenesis of autoimmune diseases of the central as well as peripheral nervous system, and we can roughly summarize the pathogenesis into three types: complement-mediated damage, alterations in receptor function (changes in receptor content, localization, and density), and changes in the levels of proteins associated with certain signaling pathways. First, complement-mediated injury plays a key role in the pathological process of GBS and NMOSD, for example, autoantibodies activate the complement system and form MAC after binding antigen in GBS in Figure 2, which in turn leads to the disappearance of voltage-gated sodium channels, which may lead to the occurrence of demyelination injury [82]. Secondly, the functional changes caused by autoantibodies in the density of receptors on the surface as well as in the localization of synapses have a great impact on the occurrence of the disease. Taking the anti-NMDAR encephalitis in Figure 1 as an example, the induced binding of anti-NMDAR antibodies to NMDAR leads to NMDAR internalization resulting in a decrease in the density of the NMDAR surface, and a decrease in the selective and reversible localization of synapses, which then leads to cognitive deficits in the patient and the development of neuropsychiatric symptoms [44,45]. Finally, in the pathogenesis of neurological diseases, autoantibody-induced alterations in the levels of signaling pathway proteins also play an important role in the generation of the disease, e.g., Figure 1, in anti-DDPX encephalitis, where DPPX is the regulatory subunit of the voltage-gated d-type Kv4.2 potassium channel complex, patient antibodies inhibit the expression of DPPX as well as kv4.2, leading to neuronal damage [74]. It is clear that the effects of autoantibodies on complement, receptor, and signaling pathway-related proteins play an important role in the pathogenesis of neurological disorders.
|
Autoantigens of the target |
Diseases |
Antibody pathogenicity |
Refs |
|
APQ4 |
NMOSD |
Complement activation, inflammatory response destroys receptors and induces p-Tau accumulation. |
[34,68] |
|
NMDAR |
Anti-NMDAR encephalitis |
NMDAR internalization, disruption of NMDAR/EphB2R receptor. |
[44-46] |
|
LGI1 |
Anti-LGI1 encephalitis |
Inhibits the interaction of LGI1 with ADAM22/23 and decreases Kv1 and AMPA receptor levels. |
[69-71] |
|
Epilepsy |
Reduced AMPA-R expression, downregulates inhibitory neuronal networks, leading to hyperexcitability of neuronal networks. |
[72] |
|
|
CASPR2 |
Anti-CASPR2 encephalitis |
Inhibits Caspr2 interaction with contactin-2. |
[73] |
|
AMPAR |
Anti-AMPAR encephalitis |
AMPAR internalizes and degrades. |
[74] |
|
DPPX |
Anti-DPPX encephalitis |
Reduced DPPX and Kv4.2 membrane expression. |
[74,75] |
|
ß2AdR, mAChR |
Myalgic encephalomyelitis/ chronic fatigue syndrome (ME/CFS) |
Receptor dysfunction, exact mechanism unknown. |
[49] |
|
GAD |
Stiffman syndrome, SMS |
Decreases GABA levels and increases nervous system excitability. |
[54,76,77] |
|
IgLON5 |
Anti-IgLON5 disease |
Disruption of the neuronal cytoskeleton induces p-tau accumulation, disruption of synapses. |
[54,60] |
|
Neurexin-3α |
Encephalitis |
Reduces the total number of synapses in developing neurons by causing a decrease in neurexin-3α specificity. |
[40,78] |
|
Autoantigens of the target |
Diseases |
Antibody pathogenicity |
Refs |
|
GM1, GD1a |
Acute motor axonal neuropathy (AMAN) |
Complement activation, MAC formation, clear of axons by macrophages. |
[81,82] |
|
Acute inflammatory demyelinating polyneuropathy (AIDP) |
Complement activation, MAC formation, macrophages invade the myelin sheath. |
||
|
Acute motor sensory axonal neuropathy (AMSAN) |
Complement activation, conduction was impaired at the nodes of Ranvier, obstruction of motor fiber axonal conduction. |
||
|
CRPS |
complex regional pain syndrome (CRPS) |
Release of inflammatory mediators, sensitive of primary sensory neurons, central sensitization was observed in the spinal dorsal horn. |
[100,101] |
|
GT1a |
Pharyngocervicobrachial variation (PCB) |
Complement activation, disruption of axonal glial or neuromuscular junctions. |
[102,103] |
|
GQ1b |
Miller-Fisher syndrome (MFS) |
Release of Ach in large and quantitative quantities, disruption of neuromuscular transmission. |
[104-106] |
|
NF155 |
Chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) |
Selective depletion of NF155 (in peripheral nerves), inhibition of glial junction formation in the paranodal axis, interference with nerve conduction. |
[87,89] |
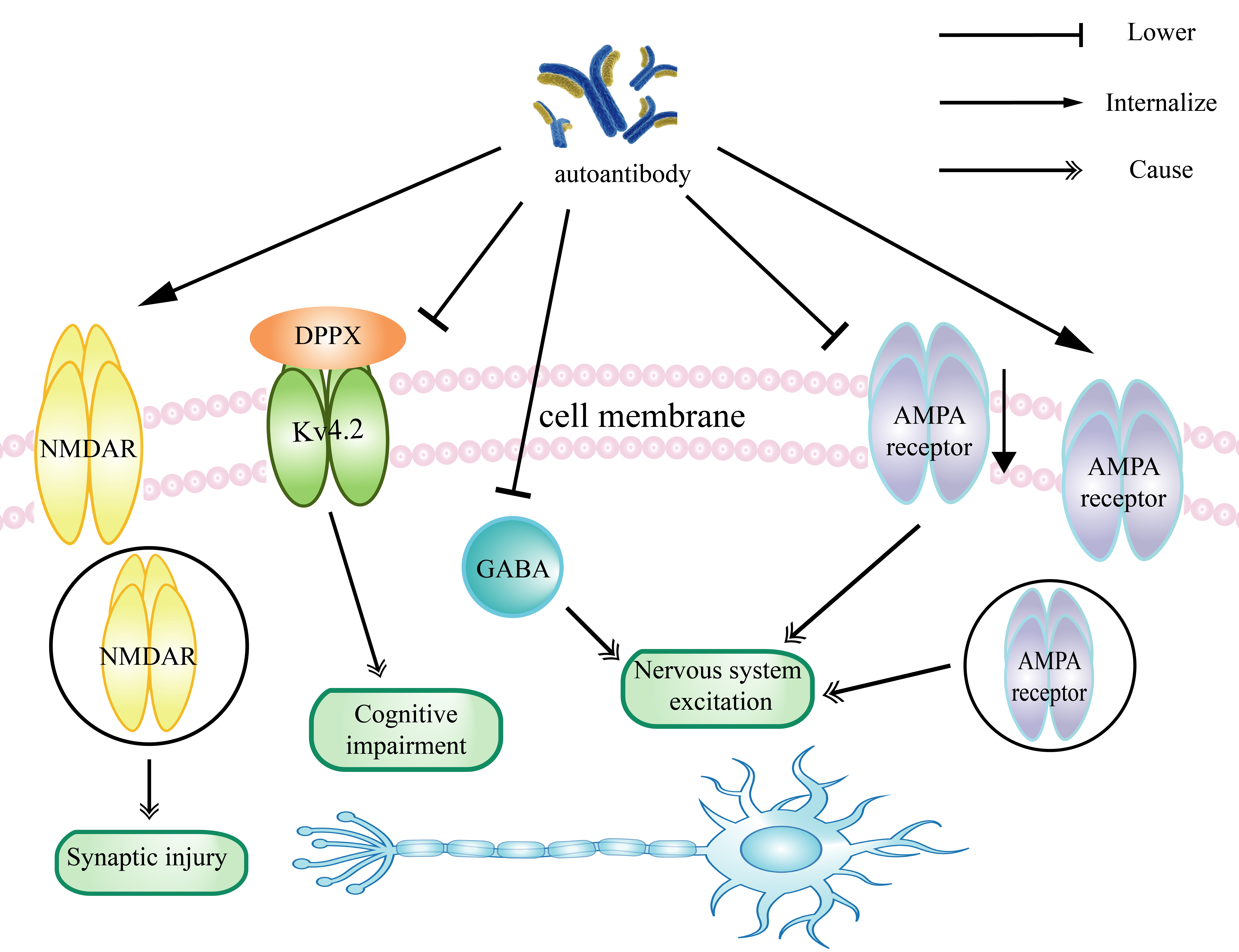
Figure 1. Mechanistic pathways of autoantibodies in the central nervous system.
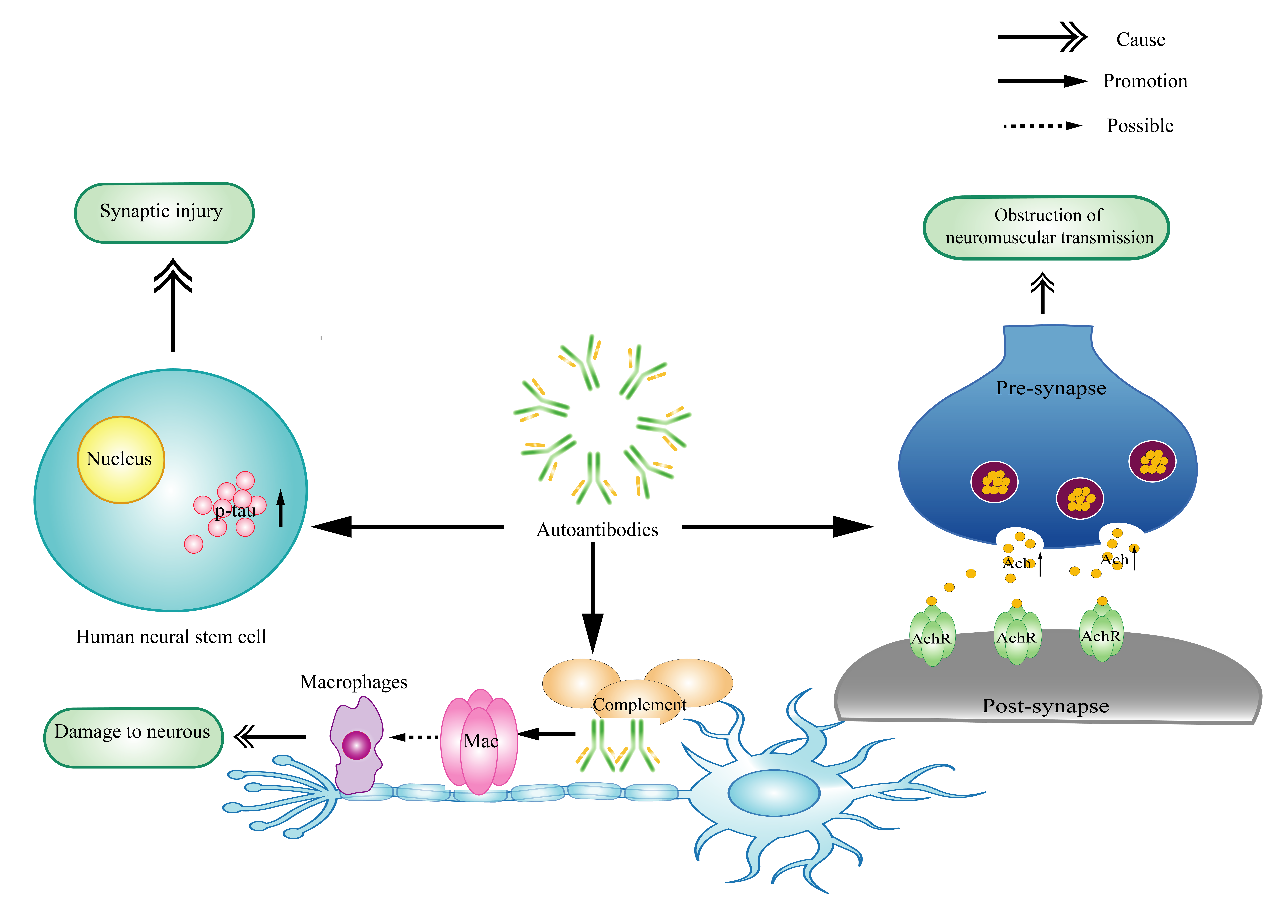
Figure 2. Mechanistic pathways of autoantibodies in the peripheral nervous system.

High-titer autoantibodies are considered to be important clinical indications of autoimmune diseases and are used as diagnostic markers in many autoimmune diseases. In recent years, many new autoantibodies have been identified in neurological-related diseases and found to play an important role in diagnosis, for example, in patients with NMOSD, serum autoantibodies against aquaporin-4 have been used as a diagnostic marker for NMOSD [107]. In glioma patients, GFAP autoantibodies, as well as anti-FLNC autoantibodies whose expression levels negatively correlate with FLNC expression, could be used as potential serum biomarkers for the early diagnosis of low-grade gliomas. In addition, some autoantibodies, although not yet identified as diagnostic markers for neurological-related diseases, have a greater potential to become clinical diagnostics, for example, in anti-IgLON5 patients, the anti-IgLON5 IgG titer in serum was positively correlated with the duration of the disease, suggesting that the expression level of IgLON5 IgG in serum may be used as one of the criteria for diagnosis [59]. In patients with CIDP, Contactin-1 and anti-NF155 antibodies also have the potential to serve as diagnostic markers of the disease by virtue of their ability to directly influence the action of key target proteins involved in the pathogenesis of CIDP [88,89]. In GBS disease, anti-GM1 and anti-GD1α antibodies can lead to disease directly through activation of complement and may also be used in the diagnosis of GBS. Autoantibodies have a very promising future as diagnostic markers in neurological-related diseases, and this idea has been presented and supported in a large body of literature [82]. In neurological-related diseases, factors such as whether autoantibodies are expressed in high titers, whether they are involved in key pathogenic mechanisms of the disease, and whether they are clearly pathogenic determine the criteria for their ability to be used as diagnostic markers.
Our lab has previously screened monoclonal autoantibody 2F5 from the peripheral blood of SLE patients for its high affinity to brain tissue proteins and identified its target protein as DDX5. In response to this, our group has prepared a fully human monoclonal autoantibody 2F5 that targets DDX5 and significantly reduces the amount of DDX5 in the nuclei of neuroglia. B-cell hyperactivation and high autoantibody production are characteristic hallmarks of SLE. The results of small RNA microarrays confirmed that 2F5 could increase the level of miR-125a-5p in B cells. Follow-up studies revealed that 2F5 significantly decreased the expression of blood-brain barrier intercellular tight junction proteins including Claudin-1, Claudin-5 and ZO-1. Tight junction is an important barrier complex between cells, which plays an important role in maintaining the homeostasis of the intracellular environment as well as in protecting the normal structure of the tissues and cells, and it is an important component of the BBB. By reviewing related literature, we confirmed that B-cell translocation gene 2 (BTG2), as a negative regulator of miR-125a-5p, plays a role in the STAT3/IL-6 pathway, and IL-6, as an important pro-inflammatory factor, may act on tight junction proteins to cause barrier damage. On this basis, the group speculated a pathway causing barrier damage from 2F5→miR-125a-5p→BTG2→STAT3/IL-6. In addition, through literature search, we have also found that many autoantibodies inactivate glial cells (mainly astrocytes) by binding to carrier channel proteins in the membrane of tight junctions, and some even inactivate glial cells directly, or inhibit the expression of tight junction proteins so that more autoantibodies, pathogens, or other harmful substances can penetrate into the BBB, thus causing CNS diseases. We hypothesize that the disruption of BBB function due to the deletion of these tight junction proteins or the opening of channels may be an important process in neuropsychiatric lupus (NPSLE), and that the specific molecular mechanisms and signaling pathways need to be explained by more research.
A large body of literature suggests that autoantibodies play an important role in immune-related neurological disorders, but research on the specific mechanism of action of certain autoantibodies is still unclear, and there is no systematic compilation of the mechanism of action of autoantibodies in neurological disorders. Without the ability to determine the causative mechanism of the disease, it is impossible to treat the patients with certainty. Therefore, accelerating the research on the role and mechanism of autoantibodies in neurological diseases is the next direction to focus on. This review explores the role and mechanism of autoantibodies in neurological diseases associated with immune abnormalities, and systematically summarizes the mechanism of autoantibodies in a series of neurological diseases, including NMOSD, anti-NMDAR encephalitis, CIDP, etc. This review will help researchers to carry out experimental studies and investigations in a more targeted manner, which will help to gain a deeper understanding of the development of the field of neuroimmunology, and at the same time will have a significant impact on the diagnosis, therapy and treatment of neurological disorders. It is important to provide guidance for the diagnosis, treatment, prevention, and control of neurological diseases. For example, anti-NF155 antibody, a marker of CIDP, may serve as a new direction for detection and treatment in future medicine. This review provides important clues to the pathogenesis of immune-related neurological diseases, not only points out the gaps in current research, but also has a certain impact on the development of autoantibody applications in neurological diseases.
Acknowledgments
This work was supported by grants from the Zhejiang Provincial Natural Science Foundation (ZJNSF) (LY20H250001), the National Natural Science Foundation of China (NSFC) (81601428), the Key Laboratory of Aging and Cancer Biology of Zhejiang Province and its open project (2020E10016), 2021 Student Innovation and Entrepreneurship Training Program Project Declaration (S202110346009), 2022 National Student Innovation and Entrepreneurship Training Program Project Funding Year National Student Innovation and Entrepreneurship Training Program (1085C5212230520), the General Scientific Research project of Education Department of Zhejiang province (Y202249995), Hangzhou Science and Technology Development Plans (20110833B29), Hangzhou Normal University School of Basic Medicine Teaching Reform and Curriculum Construction Project (JCYXYKJ202302).
References
2. Gris JC, Nobile B, Bouvier S. Neuropsychiatric presentations of antiphospholipid antibodies. Thromb Res. 2015;135 Suppl 1:S56-9.
3. Seet D, Allameen NA, Tay SH, Cho J, Mak A. Cognitive Dysfunction in Systemic Lupus Erythematosus: Immunopathology, Clinical Manifestations, Neuroimaging and Management. Rheumatol Ther. 2021;8(2):651-79.
4. Slawecki CJ, Samson HH. Changes in oral ethanol self-administration patterns resulting from ethanol concentration manipulations. Alcohol Clin Exp Res. 1997;21(6):1144-9.
5. Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11(6):535-44.
6. Pollak TA, Lennox BR, Muller S, Benros ME, Pruss H, Tebartz van Elst L, et al. Autoimmune psychosis: an international consensus on an approach to the diagnosis and management of psychosis of suspected autoimmune origin. Lancet Psychiatry. 2020;7(1):93-108.
7. Pisetsky DS. Pathogenesis of autoimmune disease. Nat Rev Nephrol. 2023;19(8):509-24.
8. Xiao ZX, Miller JS, Zheng SG. An updated advance of autoantibodies in autoimmune diseases. Autoimmun Rev. 2021;20(2):102743.
9. Nossal GJ. The double helix and immunology. Nature. 2003;421(6921):440-4.
10. Parnes O. Autoimmune disease. Lancet. 2006;367(9508):389.
11. Kang EH, Ha YJ, Lee YJ. Autoantibody Biomarkers in Rheumatic Diseases. Int J Mol Sci. 2020;21(4).
12. Lou H, Ling GS, Cao X. Autoantibodies in systemic lupus erythematosus: From immunopathology to therapeutic target. J Autoimmun. 2022;132:102861.
13. Gordon RE, Nemeth JF, Singh S, Lingham RB, Grewal IS. Harnessing SLE Autoantibodies for Intracellular Delivery of Biologic Therapeutics. Trends Biotechnol. 2021;39(3):298-310.
14. Sokolova MV, Schett G, Steffen U. Autoantibodies in Rheumatoid Arthritis: Historical Background and Novel Findings. Clin Rev Allergy Immunol. 2022;63(2):138-51.
15. Sciascia S, Bizzaro N, Meroni PL, Dimitrios B, Borghi MO, Bossuyt X, et al. Autoantibodies testing in autoimmunity: Diagnostic, prognostic and classification value. Autoimmun Rev. 2023;22(7):103356.
16. Sakuraba K, Krishnamurthy A, Sun J, Zheng X, Xu C, Peng B, et al. Autoantibodies targeting malondialdehyde-modifications in rheumatoid arthritis regulate osteoclasts via inducing glycolysis and lipid biosynthesis. J Autoimmun. 2022;133:102903.
17. Derksen V, Huizinga TWJ, van der Woude D. The role of autoantibodies in the pathophysiology of rheumatoid arthritis. Semin Immunopathol. 2017;39(4):437-46.
18. Nihei Y, Suzuki H, Suzuki Y. Current understanding of IgA antibodies in the pathogenesis of IgA nephropathy. Front Immunol. 2023;14:1165394.
19. Sethi S, De Vriese AS, Fervenza FC. Acute glomerulonephritis. Lancet. 2022;399(10335):1646-63.
20. Cavazzana I, Vojinovic T, Airo P, Fredi M, Ceribelli A, Pedretti E, et al. Systemic Sclerosis-Specific Antibodies: Novel and Classical Biomarkers. Clin Rev Allergy Immunol. 2023;64(3):412-30.
21. Chepy A, Bourel L, Koether V, Launay D, Dubucquoi S, Sobanski V. Can Antinuclear Antibodies Have a Pathogenic Role in Systemic Sclerosis? Front Immunol. 2022;13:930970.
22. Shah S, Denton CP. Scleroderma autoantibodies in guiding monitoring and treatment decisions. Curr Opin Rheumatol. 2022;34(6):302-10.
23. Baumann R, Blass C, Gotz R, Dragon S. Ontogeny of catecholamine and adenosine receptor-mediated cAMP signaling of embryonic red blood cells: role of cGMP-inhibited phosphodiesterase 3 and hemoglobin. Blood. 1999;94(12):4314-20.
24. Satoh M, Ceribelli A, Hasegawa T, Tanaka S. Clinical Significance of Antinucleolar Antibodies: Biomarkers for Autoimmune Diseases, Malignancies, and others. Clin Rev Allergy Immunol. 2022;63(2):210-39.
25. Zhu Q, He P, Zheng C, Chen Z, Qi S, Zhou D, et al. Identification and evaluation of novel serum autoantibody biomarkers for early diagnosis of gastric cancer and precancerous lesion. J Cancer Res Clin Oncol. 2023;149(11):8369-78.
26. Qin R, Wu H, Guan H, Tang C, Zheng Z, Deng C, et al. Anti-phospholipid autoantibodies in human diseases. Clin Immunol. 2023;256:109803.
27. Miyashita K, Lutz J, Hudgins LC, Toib D, Ashraf AP, Song W, et al. Chylomicronemia from GPIHBP1 autoantibodies. J Lipid Res. 2020;61(11):1365-76.
28. Beigneux AP, Miyashita K, Ploug M, Blom DJ, Ai M, Linton MF, et al. Autoantibodies against GPIHBP1 as a Cause of Hypertriglyceridemia. N Engl J Med. 2017;376(17):1647-58.
29. Altmann DM, Whettlock EM, Liu S, Arachchillage DJ, Boyton RJ. The immunology of long COVID. Nat Rev Immunol. 2023;23(10):618-34.
30. Group GBDNDC. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16(11):877-97.
31. Collaborators GBDN. Global, regional, and national burden of neurological disorders, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459-80.
32. Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, et al. Trial of Satralizumab in Neuromyelitis Optica Spectrum Disorder. N Engl J Med. 2019;381(22):2114-24.
33. Shimizu F, Schaller KL, Owens GP, Cotleur AC, Kellner D, Takeshita Y, et al. Glucose-regulated protein 78 autoantibody associates with blood-brain barrier disruption in neuromyelitis optica. Sci Transl Med. 2017;9(397):eaai9111.
34. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473-7.
35. Dejanovic B, Huntley MA, De Maziere A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron. 2018;100(6):1322-36e7.
36. Howe CL, Kaptzan T, Magana SM, Ayers-Ringler JR, LaFrance-Corey RG, Lucchinetti CF. Neuromyelitis optica IgG stimulates an immunological response in rat astrocyte cultures. Glia. 2014;62(5):692-708.
37. Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH, et al. Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest. 2020;130(8):4025-38.
38. Liszewski MK, Kolev M, Le Friec G, Leung M, Bertram PG, Fara AF, et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity. 2013;39(6):1143-57.
39. Tzang RF, Chang CH, Chang YC, Lane HY. Autism Associated With Anti-NMDAR Encephalitis: Glutamate-Related Therapy. Front Psychiatry. 2019;10:440.
40. Dalmau J, Armangue T, Planaguma J, Radosevic M, Mannara F, Leypoldt F, et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019;18(11):1045-57.
41. Kayser MS, Dalmau J. Anti-NMDA receptor encephalitis, autoimmunity, and psychosis. Schizophr Res. 2016;176(1):36-40.
42. Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14(6):383-400.
43. Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8(6):413-26.
44. Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, et al. Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci. 2010;30(17):5866-75.
45. Shu Y, Peng F, Zhao B, Liu C, Li Q, Li H, et al. Transfer of patient's peripheral blood mononuclear cells (PBMCs) disrupts blood-brain barrier and induces anti-NMDAR encephalitis: a study of novel humanized PBMC mouse model. J Neuroinflammation. 2023;20(1):164.
46. Reincke SM, von Wardenburg N, Homeyer MA, Kornau HC, Spagni G, Li LY, et al. Chimeric autoantibody receptor T cells deplete NMDA receptor-specific B cells. Cell. 2023;186(23):5084-97 e18.
47. Sotzny F, Blanco J, Capelli E, Castro-Marrero J, Steiner S, Murovska M, et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome - Evidence for an autoimmune disease. Autoimmun Rev. 2018;17(6):601-9.
48. Freitag H, Szklarski M, Lorenz S, Sotzny F, Bauer S, Philippe A, et al. Autoantibodies to Vasoregulative G-Protein-Coupled Receptors Correlate with Symptom Severity, Autonomic Dysfunction and Disability in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J Clin Med. 2021;10(16):3675.
49. Wirth K, Scheibenbogen C. A Unifying Hypothesis of the Pathophysiology of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS): Recognitions from the finding of autoantibodies against ss2-adrenergic receptors. Autoimmun Rev. 2020;19(6):102527.
50. Wirth KJ, Scheibenbogen C. Pathophysiology of skeletal muscle disturbances in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome (ME/CFS). J Transl Med. 2021;19(1):162.
51. Menestrina F, Novelli P, Bonetti F, Iannucci AM. [Pericardial teratoma: case report (author's transl)]. Pathologica. 1980;72(1020):589-95.
52. Wei P, Zhang W, Yang LS, Zhang HS, Xu XE, Jiang YH, et al. Serum GFAP autoantibody as an ELISA-detectable glioma marker. Tumour Biol. 2013;34(4):2283-92.
53. Dade M, Berzero G, Izquierdo C, Giry M, Benazra M, Delattre JY, et al. Neurological Syndromes Associated with Anti-GAD Antibodies. Int J Mol Sci. 2020;21(10):3701.
54. Baizabal-Carvallo JF. The neurological syndromes associated with glutamic acid decarboxylase antibodies. J Autoimmun. 2019;101:35-47.
55. Arino H, Hoftberger R, Gresa-Arribas N, Martinez-Hernandez E, Armangue T, Kruer MC, et al. Paraneoplastic Neurological Syndromes and Glutamic Acid Decarboxylase Antibodies. JAMA Neurol. 2015;72(8):874-81.
56. Maimaiti B, Mijiti S, Sun H, Xie Y, Jiang T, Meng Q, et al. Are anti-glutamic acid decarboxylase 65-kDa isoform antibodies related to diabetes or brain tumor? Eur J Med Res. 2022;27(1):53.
57. Adachi-Hayama M, Adachi A, Shinozaki N, Matsutani T, Hiwasa T, Takiguchi M, et al. Circulating anti-filamin C autoantibody as a potential serum biomarker for low-grade gliomas. BMC Cancer. 2014;14:452.
58. Landa J, Gaig C, Plaguma J, Saiz A, Antonell A, Sanchez-Valle R, et al. Effects of IgLON5 Antibodies on Neuronal Cytoskeleton: A Link between Autoimmunity and Neurodegeneration. Ann Neurol. 2020;88(5):1023-7.
59. Gruter T, Mollers FE, Tietz A, Dargvainiene J, Melzer N, Heidbreder A, et al. Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease. Brain. 2023;146(2):600-11.
60. Zhang YH, Ni Y, Gao YN, Shen DD, He L, Yin D, et al. Anti-IgLON5 disease: a novel topic beyond neuroimmunology. Neural Regen Res. 2023;18(5):1017-22.
61. Kuijf ML, Samsom JN, van Rijs W, Bax M, Huizinga R, Heikema AP, et al. TLR4-mediated sensing of Campylobacter jejuni by dendritic cells is determined by sialylation. J Immunol. 2010;185(1):748-55.
62. Bing-Lei W, Jia-Hua Z, Yan L, Zan Y, Xin B, Jian-Hua S, et al. Three cases of antibody-LGI1 limbic encephalitis and review of literature. Int J Neurosci. 2019;129(7):642-8.
63. Bonday ZQ, Dhanasekaran S, Rangarajan PN, Padmanaban G. Import of host delta-aminolevulinate dehydratase into the malarial parasite: identification of a new drug target. Nat Med. 2000;6(8):898-903.
64. Fels E, Muniz-Castrillo S, Vogrig A, Joubert B, Honnorat J, Pascual O. Role of LGI1 protein in synaptic transmission: From physiology to pathology. Neurobiol Dis. 2021;160:105537.
65. Kjaergaard JJ, Salling N, Magid E, Ditzel J. Serum amylase during recovery from diabetic ketoacidosis. Diabete Metab. 1984;10(1):25-30.
66. Ambudkar SV, Maloney PC. Anion exchange in bacteria: reconstitution of phosphate: hexose 6-phosphate antiport from Streptococcus lactis. Methods Enzymol. 1986;125:558-63.
67. Grosvenor PW, Gray DO. Colutequinone and colutehydroquinone, antifungal isoflavonoids from Colutea arborescens. Phytochemistry. 1996;43(2):377-80.
68. Tice C, McDevitt J, Langford D. Astrocytes, HIV and the Glymphatic System: A Disease of Disrupted Waste Management? Front Cell Infect Microbiol. 2020;10:523379.
69. Govert F, Abrante L, Becktepe J, Balint B, Ganos C, Hofstadt-van Oy U, et al. Distinct movement disorders in contactin-associated-protein-like-2 antibody-associated autoimmune encephalitis. Brain. 2023;146(2):657-67.
70. Lai M, Huijbers MG, Lancaster E, Graus F, Bataller L, Balice-Gordon R, et al. Investigation of LGI1 as the antigen in limbic encephalitis previously attributed to potassium channels: a case series. Lancet Neurol. 2010;9(8):776-85.
71. Seery N, Butzkueven H, O'Brien TJ, Monif M. Contemporary advances in antibody-mediated encephalitis: anti-LGI1 and anti-Caspr2 antibody (Ab)-mediated encephalitides. Autoimmun Rev. 2022;21(5):103074.
72. Fels E, Mayeur ME, Wayere E, Vincent C, Malleval C, Honnorat J, et al. Dysregulation of the hippocampal neuronal network by LGI1 auto-antibodies. PLoS One. 2022;17(8):e0272277.
73. Patterson KR, Dalmau J, Lancaster E. Mechanisms of Caspr2 antibodies in autoimmune encephalitis and neuromyotonia. Ann Neurol. 2018;83(1):40-51.
74. Seery N, Butzkueven H, O'Brien TJ, Monif M. Rare antibody-mediated and seronegative autoimmune encephalitis: An update. Autoimmun Rev. 2022;21(7):103118.
75. Hara M, Arino H, Petit-Pedrol M, Sabater L, Titulaer MJ, Martinez-Hernandez E, et al. DPPX antibody-associated encephalitis: Main syndrome and antibody effects. Neurology. 2017;88(14):1340-8.
76. Graus F, Saiz A, Dalmau J. GAD antibodies in neurological disorders - insights and challenges. Nat Rev Neurol. 2020;16(7):353-65.
77. Hansen N, Krasiuk I, Titsch T. Neural autoantibodies in delirium. J Autoimmun. 2021;125:102740.
78. Gresa-Arribas N, Planaguma J, Petit-Pedrol M, Kawachi I, Katada S, Glaser CA, et al. Human neurexin-3alpha antibodies associate with encephalitis and alter synapse development. Neurology. 2016;86(24):2235-42.
79. Koike H, Katsuno M. Macrophages and Autoantibodies in Demyelinating Diseases. Cells. 2021;10(4):844.
80. Seixas AI, Morais MRG, Brakebusch C, Relvas JB. A RhoA-mediated biomechanical response in Schwann cells modulates peripheral nerve myelination. Prog Neurobiol. 2023;227:102481.
81. Islam Z, Jacobs BC, van Belkum A, Mohammad QD, Islam MB, Herbrink P, et al. Axonal variant of Guillain-Barre syndrome associated with Campylobacter infection in Bangladesh. Neurology. 2010;74(7):581-7.
82. van den Berg B, Walgaard C, Drenthen J, Fokke C, Jacobs BC, van Doorn PA. Guillain-Barre syndrome: pathogenesis, diagnosis, treatment and prognosis. Nat Rev Neurol. 2014;10(8):469-82.
83. Hughes RA, Cornblath DR. Guillain-Barre syndrome. Lancet. 2005;366(9497):1653-66.
84. Davidson AI, Halstead SK, Goodfellow JA, Chavada G, Mallik A, Overell J, et al. Inhibition of complement in Guillain-Barre syndrome: the ICA-GBS study. J Peripher Nerv Syst. 2017;22(1):4-12.
85. Doets AY, Hughes RA, Brassington R, Hadden RD, Pritchard J. Pharmacological treatment other than corticosteroids, intravenous immunoglobulin and plasma exchange for Guillain-Barre syndrome. Cochrane Database Syst Rev. 2020;1(1):CD008630.
86. Florian IA, Lupan I, Sur L, Samasca G, Timis TL. To be, or not to be... Guillain-Barre Syndrome. Autoimmun Rev. 2021;20(12):102983.
87. Koike H, Kadoya M, Kaida KI, Ikeda S, Kawagashira Y, Iijima M, et al. Paranodal dissection in chronic inflammatory demyelinating polyneuropathy with anti-neurofascin-155 and anti-contactin-1 antibodies. J Neurol Neurosurg Psychiatry. 2017;88(6):465-73.
88. Vallat JM, Yuki N, Sekiguchi K, Kokubun N, Oka N, Mathis S, et al. Paranodal lesions in chronic inflammatory demyelinating polyneuropathy associated with anti-Neurofascin 155 antibodies. Neuromuscul Disord. 2017;27(3):290-3.
89. Manso C, Querol L, Lleixa C, Poncelet M, Mekaouche M, Vallat JM, et al. Anti-Neurofascin-155 IgG4 antibodies prevent paranodal complex formation in vivo. J Clin Invest. 2019;129(6):2222-36.
90. Hu B, Wang C, Chang Q, Yang W, Wu Z, Meng M, et al. NF155-overexpression promotes remyelination and functional restoration in a hypoxic-ischemic mixed neonatal rat forebrain cell culture system. Neurosci Lett. 2020;718:134743.
91. Tang L, Huang Q, Qin Z, Tang X. Distinguish CIDP with autoantibody from that without autoantibody: pathogenesis, histopathology, and clinical features. J Neurol. 2021;268(8):2757-68.
92. Querol L, Crabtree M, Herepath M, Priedane E, Viejo Viejo I, Agush S, et al. Systematic literature review of burden of illness in chronic inflammatory demyelinating polyneuropathy (CIDP). J Neurol. 2021;268(10):3706-16.
93. Shahrizaila N, Yuki N. Bickerstaff brainstem encephalitis and Fisher syndrome: anti-GQ1b antibody syndrome. J Neurol Neurosurg Psychiatry. 2013;84(5):576-83.
94. Chiba A, Kusunoki S, Obata H, Machinami R, Kanazawa I. Serum anti-GQ1b IgG antibody is associated with ophthalmoplegia in Miller Fisher syndrome and Guillain-Barre syndrome: clinical and immunohistochemical studies. Neurology. 1993;43(10):1911-7.
95. Liu JX, Willison HJ, Pedrosa-Domellof F. Immunolocalization of GQ1b and related gangliosides in human extraocular neuromuscular junctions and muscle spindles. Invest Ophthalmol Vis Sci. 2009;50(7):3226-32.
96. Jacobs BC, O'Hanlon GM, Bullens RW, Veitch J, Plomp JJ, Willison HJ. Immunoglobulins inhibit pathophysiological effects of anti-GQ1b-positive sera at motor nerve terminals through inhibition of antibody binding. Brain. 2003;126(Pt 10):2220-34.
97. Chen HJ, Lee TC, Wei CP. Treatment of cerebellar infarction by decompressive suboccipital craniectomy. Stroke. 1992;23(7):957-61.
98. O'Hanlon GM, Humphreys PD, Goldman RS, Halstead SK, Bullens RW, Plomp JJ, et al. Calpain inhibitors protect against axonal degeneration in a model of anti-ganglioside antibody-mediated motor nerve terminal injury. Brain. 2003;126(Pt 11):2497-509.
99. Overell JR, Willison HJ. Recent developments in Miller Fisher syndrome and related disorders. Curr Opin Neurol. 2005;18(5):562-6.
100. Cuhadar U, Gentry C, Vastani N, Sensi S, Bevan S, Goebel A, et al. Autoantibodies produce pain in complex regional pain syndrome by sensitizing nociceptors. Pain. 2019;160(12):2855-65.
101. Goebel A, Blaes F. Complex regional pain syndrome, prototype of a novel kind of autoimmune disease. Autoimmun Rev. 2013;12(6):682-6.
102. Koga M, Yoshino H, Morimatsu M, Yuki N. Anti-GT1a IgG in Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2002;72(6):767-71.
103. Wakerley BR, Yuki N. Pharyngeal-cervical-brachial variant of Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 2014;85(3):339-44.
104. Goodyear CS, O'Hanlon GM, Plomp JJ, Wagner ER, Morrison I, Veitch J, et al. Monoclonal antibodies raised against Guillain-Barre syndrome-associated Campylobacter jejuni lipopolysaccharides react with neuronal gangliosides and paralyze muscle-nerve preparations. J Clin Invest. 1999;104(6):697-708.
105. Plomp JJ, Molenaar PC, O'Hanlon GM, Jacobs BC, Veitch J, Daha MR, et al. Miller Fisher anti-GQ1b antibodies: alpha-latrotoxin-like effects on motor end plates. Ann Neurol. 1999;45(2):189-99.
106. Roberts M, Willison H, Vincent A, Newsom-Davis J. Serum factor in Miller-Fisher variant of Guillain-Barre syndrome and neurotransmitter release. Lancet. 1994;343(8895):454-5.
107. Kuwabara S, Yuki N, Koga M, Hattori T, Matsuura D, Miyake M, et al. IgG anti-GM1 antibody is associated with reversible conduction failure and axonal degeneration in Guillain-Barre syndrome. Ann Neurol. 1998;44(2):202-8.