Abstract
There is a myriad of potential candidates suggested to be associated to the aetiology of Parkinson’s disease, of these a-synuclein appears to the first runner up. This notion is endorsed by the appearance of a-synuclein aggregates (present in Lewy bodies) in close proximity to regions exhibiting neuronal cell loss. The mechanism(s) contributing to its accumulation include, some genetic dysfunction, overproduction of the protein, misfolding, inability to effectively degrade the misfolded form. Furthermore, the ability of a-synuclein to operate as ferrireductase, allows it to produce reactive oxygen species and exacerbate the oxidative stress related cytotoxic processes in the illness. The accumulation of misfolded a-synuclein may also invoke the release of inflammatory mediators. The presence of microgliosis in the substantia nigra in the disease reflects the occurrence of inflammation. The destructive role of neuro-inflammation in degenerative diseases is exhibited by the neurological manifestations produced by infectious agents such as bacteria and viruses. Therefore, these findings offer many budding therapeutic interventions that can be advocated in the disorder. However, an urgent need for a biomarker is warranted to detect the disease early in order to halt or delay its progression.
Keywords
Alpha-synuclein, Microglia, Neuroinflammation, Substantia nigra, Neuronal death, Parkinson’s disease
Short Communication
The neurodegenerative disorder Parkinson’s disease (PD) is hallmarked by the presence of cytoplasmic inclusions, Lewy body (LB) and ravaged dopaminergic neurons in the substantia nigra pars compacta (SNc). LB are primarily composed of aggregates of the protein α-synuclein. Although the precise aetiology underlying the illness remains obscure, a medley of factors has been postulated. There are a multitude of factors that have been implicated including, ageing, genetic predisposition, endogenous/ exogenous toxins, iron, oxidative stress, mitochondrial dysfunction, misfolding of proteins (such as α-synuclein), inefficient clearance of rogue proteins, infectious agents (such as viruses) and neuroinflammation. This list is not exhaustive and current findings suggest other evolving influences. More importantly, it is highly likely that rather than operating independently, these formidable culprits intermesh to invoke cytotoxic processes resulting in severe neuronal causalities in its wake. This concords with the notion of a multifactorial aetiology for PD [1].
In the event of a neurodegenerative illness or infection, microglia is activated. This idea is supported by the presence of HLA-DR- positive or class II major histocompatibility (MHC class II antigen) reactive microgliosis in SN in PD [2]. Similar findings were reported from in vivo PET imaging in PD patients [3]. These results may be significant to the pathogenesis, since microglia constitute to the major antigen-presenting cell during neurodegeneration. This phenomenon is demonstrated by the presence and propagation of activated microglia in other diseases such as Alzheimer’s disease, amyotrophic lateral sclerosis and prion disease [4]. So, it appears to be a part of a common and shared pathway leading to neurodegeneration. This is indicative of the occurrence of neuro-inflammatory processes, since under physiological conditions the microglia cells express low levels of MHC II molecules, thereby suggestive of an ongoing degenerative process during the illness (Figure 1). It supports the notion that the immune responses may execute an integral role in the development and/or progression of the pathophysiology. This notion is supported by the reduction of risk or delay in progression of PD by the administration of the nonsteroidal anti-inflammatory drug, ibuprofen [5,6]. More importantly, transcriptomic studies in these diseases illustrated an upregulation of genes for HLA-DR [7], which suggests that the inflammation feature observed in these diseases is derived from HLA-DR genetic predilection.
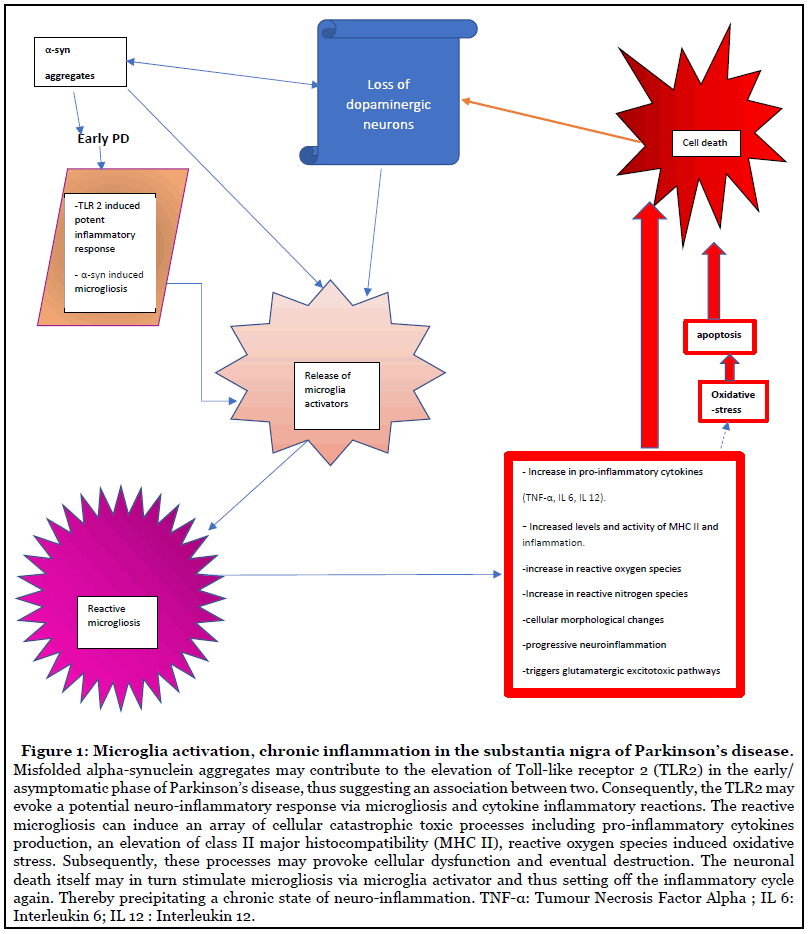
At present it is unclear whether microgliosis is a component of the primary artillery or a manifestation of the degenerative processes, nevertheless it very likely contributes to the resulting cellular carnage. Microglia can operate as either neuroprotective or destructive agents [8]. Thus, it is possible that it exerts both a malevolent and protective role. Indeed, microglia activated by lipopolysaccharide or TNF-α, are denoted as the “M1” type, that is pro-inflammatory and plays a central role in the defence against pathogens and tumour cells via the production of cytokines (including, TNF-α, IL-6 or IL- 12) and reactive free radicals. In contrast, the “M2” type is neuroprotective by virtue of its actions such as release of anti-inflammatory cytokines (such as, IL-4, IL-13 or TGF-β) and stimulates angiogenesis [9].
Perhaps the initial consequence of microgliosis is neuroprotective, since it augments phagocytosis and the clearance of misfolded proteins such as α-synuclein. Thus, they endeavour to maintain neuro-immunological homeostasis to protect the cells. It is also relatively selective, since activation of microglia and consequent reactive processes may be customised to different trigger pathological events or rogue molecules . For instance, zinc released from neurons (probably synaptic vesicles), in animal models of cerebral ischaemia provokes microglia activation [10].
However, subsequently a chronic state of inflammation (Figure 1) may result in the release of cytokines from the activated microglia , which may elicit cytotoxic proinflammatory pathway(s). Indeed, in vitro studies suggest that pro-inflammatory cytokines can induce MHC class II antigens present on microglia. These findings concord with the elevation of cytokines, potent regulators of cell growth and apoptosis-signalling receptor molecule, TNF alpha, IL-1β, IL-12,IL-6, sFAS, EGF, TGF-alpha, TGF-β1, bFGF, β2-MG) found in the striatum in PD [11-16]. Thus, cytokine release may represent a potential target site for drug treatment in PD and they may delay the degenerative processes and thus the progression. Additionally, a dual role for microgliosis has also been hypothesized in Alzheimer’ disease [17].
Exposure to the neurotoxin 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine (MPTP) produces irreversible dopamine neuronal destruction and parkinsonian syndrome in man and primates. Interestingly, subjects in contact with MPTP showed microgliosis to be maintained for around 16 years after exposure. This is suggestive of a chronic state of neuroinflammation and also highlights its relevance to the idiopathic form of PD [18]. Furthermore, animal studies using neurotoxin such as MPTP and 6-hydroxydopamine [19], suggest that microglia activation is stimulated by the initial destruction of dopamine cells, thereby representing a secondary effect. However, lipopolysaccharide stimulated microglia (probably of the M1 type) have also shown to exert cytotoxic actions on the dopaminergic neurons in both in vivo and in vitro studies [20]. Also, the T cells may conspire with the active microglia to execute neuro-destruction. In turn, the cell destruction may result in stimulation of additional immune reactions and cascade greater destruction of the neuron population, thereby endorsing the ambivalent role of the activated microglia and the close association of inflammatory organisation to neuronal death.
Neuro-inflammation appears to be a promising contender in the fabric of degenerative processes in PD particularly since the nigral dopaminergic neurons are particularly susceptible to the cytotoxic processes related to oxidative stress and neuro-inflammation. This is supported by the presence of inflammation in the SN in PD, where there is marked cell loss. Interestingly, toll-like receptor 2 (TLR2) is markedly expressed in reactive microglia in the asymptomatic phase of PD, incidental Lewy body disease (Figure 1). TLR2 facilitates α-synuclein mediated activation of microglia. However, this phenomenon is not observed in PD, thereby reflecting an early pathological reaction(s) when the dopaminergic neuronal loss is less marked since, incidental Lewy body disease exhibits around 40% nigral cell loss in contrast to 82% loss in the symptomatic PD [21]. This suggests, firstly that microgliosis is not only provoked by dopaminergic neuronal death but also by TLR2 linked α-synuclein and secondly an involvement of inflammatory processes in the initial stages of the disorder. Additionally, perhaps some genetic defects may ascribe for the malevolent role adopted by the microglia. Indeed, mutations in microglia have contribute to marked behavioural syndromes in mice.
Subsequently, as the disease progresses and there is more marked neuronal loss this may augment the cytotoxic contribution from the inflammatory system. There may be a genetic component related to the faulty or dysfunctional immunological dynamics, so exposure to infectious mediators may initiate a cascade of cytotoxic events, which may be linked to the misfolding and aggregation of α-synuclein [22]. Alternatively, α-synuclein may be frontline candidate in the neuronal destructive force [23].
The physiological role of α-synuclein includes participation in the release of neurotransmitters and more importantly in synaptic plasticity [24]. It appears to exist in unfolded monomeric (or tetrameric) form. However, under pathological conditions it alters from soluble to insoluble fibrillary structures chiefly contributing to misfolding of the protein. Subsequently, accumulation of α-synuclein appears to give rise to pathological LB structures. The presence of these neuronal inclusions in the SN is vital for diagnosis of PD at post-mortem [25], although they are also found in other neurodegenerative disorders. In view of the occurrence of LB in close vicinity to areas exhibiting marked neuronal loss, coupled with its presence in early asymptomatic PD or incidental Lewy body disease [25], lends support to the concept that α-synuclein may be lead character in the pathogenesis of PD [26]. Indeed, in accordance to Braak [27] proposed scheme for staging PD, two vital points are highlighted , firstly that α-synuclein pathology is present in the initial stages (stage 1 in the vagus nerve and olfactory nucleus) and secondly during stage 3 α-synuclein appears in the substantia nigra and this is associated with the manifestation of early motor deficits. There is an absence of α-synuclein pathology when there is only a marginal 12% nigral cell loss, whereas the appearance of α-synuclein aggregates in the neuromelanin containing SN demonstrated a significant correlation to dopaminergic neuronal death. Perhaps the amasses of α-synuclein prompts cellular cytotoxic chaos in these vulnerable dopaminergic nigral neuronal functioning resulting in their destruction. In addition, mutations of α-synuclein gene has been linked to the familial PD [28].
A genetic variability in α-synuclein has been implicated in idiopathic PD. Indeed, its accumulation has been attributed to the α-synuclein locus triplication [29]. This genetically driven aggregation of α-synuclein may activate microglia in an attempt to degrade the α-synuclein accumulation and precipitate neuro-inflammation (Figure 2). Indeed, microglial astrocytes and neurons have the potential to breakdown the accumulated α-synuclein by internal degradative mechanisms [30]. Under physiological conditions, macrophages break down α-synuclein via lysosomal and proteolytic pathways, however perhaps in the pathological state these degradative mechanisms are compromised or overwhelmed by the excess manifestation of these oligomers. It seems plausible that α-synuclein aggregates initiate neuro-inflammatory mechanisms (adaptive immunity).
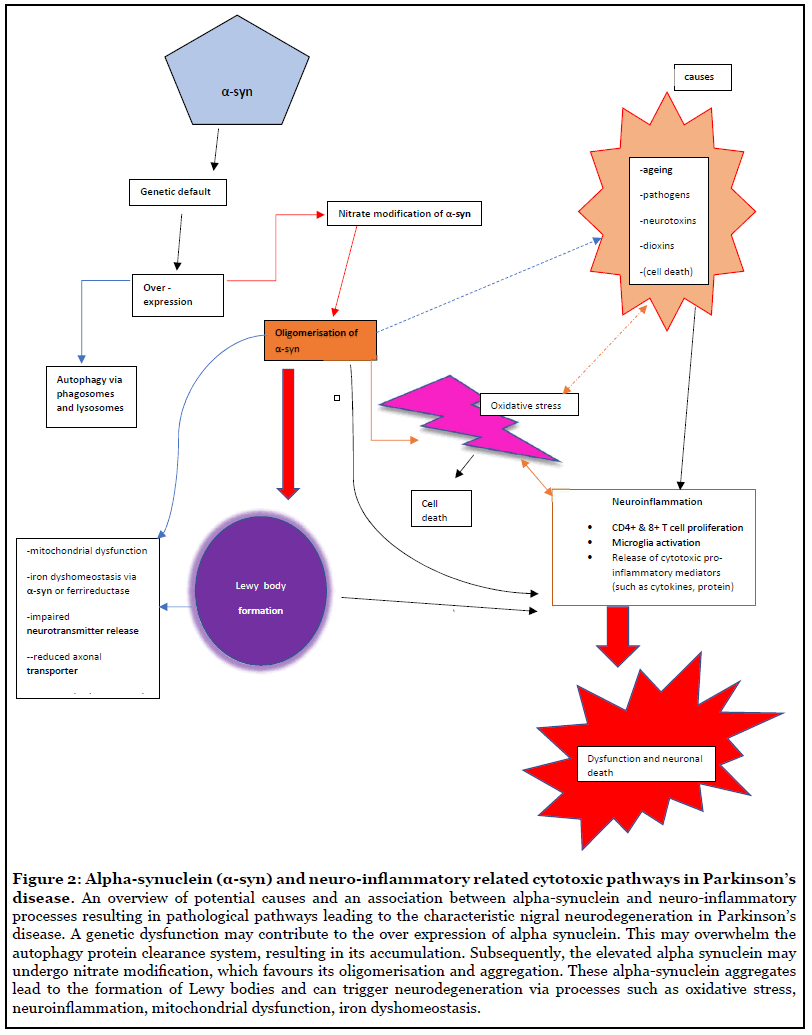
Over expression of the native form of α-synuclein lends a predilection to its misfolding (Figure 2), accumulation and subsequent formation LB [31]. This may be attributed for the tendency for the native or wild type α-synuclein to nitrate modification which augments its susceptibility to aggregate [32]. The nitro-tyrosine modified α-synuclein elicits T lymphocytes proliferation and neuronal destruction. Indeed, specific peptides on α-synuclein are recognised by T cells which provoke its cellular destructive reactions [33]. The nitrated form of α-synuclein was detected in MPTP treated mice, and interestingly this form of the protein generated T cells coupled with neuroinflammation and exacerbated destruction of dopamine cells. Similarly, an infiltration of CD4+ and CD8+T cells (Figure 2) was found in the PD brain [34]. The cytotoxic T cell actions observed in the dopaminergic neurons in PD was chiefly orchestrated by CD4+ T cell associated with Fas ligand. This notion is supported by the buildup of CD4+ T cells in protracted neurodegeneration. It therefore appears that α-synuclein not only stimulates neuro-inflammation via T cells of the adaptive immune system, but also maintains it [35]. In addition, since the B cells are often reported to be reduced in illness, humoral immunity may also been involved in pathogenesis of PD. Alternatively, there is a possibility of that an earlier affiliation or encounter with an infectious agent such as a bacteria or virus, which may somehow impact the immune system conferring to the susceptibility to induce neurodegeneration.
In the early stages of the illness peripheral inflammation appears to be entangled in the initiation and manifestation of non-motor symptoms (Braak stages 1-2). The bacteria Helicobacter pylori induced gut infections have been related to the progression of PD. This idea is supported by the motor improvement observed in PD patients after treatment for Helicobacter pylori [36]. It therefore appears chronic gut related infections may signal inflammation (via pro-inflammatory mediators) via the gut-brain axis to the brain and fund the progression of degeneration by deploying inflammatory troops such as cytotoxic cytokines.
Indeed, the overexpression of the gram-negative bacteria Proteus mirabilis in PD mouse model, exhibited destruction of dopamine neurons and inflammation in the SN coupled to the presence of α-synuclein aggregates in the brain and gastro-intestinal tract [37].
Many patients afflicted with encephalitis lethargica (1915- 1927), exhibited parkinsonian extrapyramidal like features during the chronic phase of the infection that occurred months or years later [38-42]. However, viral antibodies, RNA, viral particles or inclusions could not be detected in many studies [43-48]. These data are in contrast to those reported by others [49-51]. This idea is supported by the association of influenza (and other viral infections) to post-encephalitic parkinsonism. A positive correlation was demonstrated between severity of influenza and the occurrence of PD in a population-based study [52]. Interestingly in viral infections cytotoxic CD4+ T cells have been reported to exert a key part in the observed immune reactions. In a study they found that exposure to dioxin compromised the cytotoxic T cell response against the influenza A virus in mice [53]. Collectively, these findings are indicative of the operation of a toxin or “an infectious agent’’, which causes the immune system to turn hostile against the nigral neurons. However, α -synuclein containing Lewy bodies have not been demonstrated in post mortem brain tissue of postencephalitic parkinsonism [54]. This opens the possibility of many potential candidates in the pathogenesis of PD including environmental toxins such as dioxin, pesticides, virus, bacteria, etc. The presence of these entities coupled with neuronal cell vulnerability related to aging and genetic predisposition probably prompts the abnormal regulation of inflammatory reactions, this may in turn exacerbate neuronal destruction observed in PD.
Experimental long-term studies with H5N1 influenza virus show that H5N1 induces transient loss of dopamine in the SNpc and basal ganglia. In addition, activated microglia and increase in cytokines could be detected suggesting, that viral infection is not excluded as trigger for Parkinson’s disease [17]. Acute onset of West Nile virus infections induces parkinsonian like symptoms associated to the loss of dopaminergic neurons in the SN and an elevation of α-synuclein. The α-synuclein appears to exert a cellular protective role since it inhibits RNA viral infection associated injury [55]. Unfortunately, there are only few reports on viral brain infection in contrast to those on the degenerating potential of HIV. Recent work related to molecular pathology of Neuro-AIDS/ CNS-HIV, has clearly shown neuritic α -synuclein in the SN of 16% HIV cases and β-amyloid deposits in most of the HIV positive cases [56,57]. Mackiewicz and colleagues [57] suggested that there may be a common pathogenic mechanisms between HIV and aging, which may promote pathways involved in Alzheimer’s disease or Alzheimer related dementias including triggering β-amyloid, Tau- or α -synuclein pathology.
Post-mortem findings have demonstrated a significant decrease of the numerical density of total neurons, as well as of pigmented neurons in the SN of patients with HIV-1 infection [58]. For modelling HIV-1 long term symptomology, rhesus monkeys infected with the simian immunodeficiency virus (SIV mac 251) were employed and these studies showed the dopaminergic system to be markedly affected with an increase in dopamine turnover [59]. Using HIV-infected T-lymphoblasts ACH-2 to elucidate the effects of HIV on dopamine, a dopamine induced concentration-dependent HIV activation was reported [60]. Glutamatergic pathology (Figure 1) is of major interest in HIV induced dementia [61]. It has been suggested that the NMDA-receptor channel antagonists’ amantadine and memantine may be a suitable therapeutic option for both parkinsonism and dementia observed in late phases of HIV infection [62].
In addition, primary macrophages, neuroblastoma and glioblastoma cell lines exposed to HIV-1 indicated the operation of oxidative stress as the mechanism of degenerative processes involved in HIV-1 infection [59]. However, others have suggested that impaired repair mechanisms due to a deficiency of S-adenosylmethionine facilitates cytokine and/or oxygen mediated toxicity underlying vascular myelinopathy and neuronal damage in AIDS [63]. Furthermore, it has been shown that the DAT 10/10-repeat allele T3 is more frequent in HIV individuals. Subjects homozygous for the 10-repeat allele had higher amounts of CSF dopamine and reduced DATmRNA expression but similar disease severity compared to those carrying other DAT genotypes [64].
Recently, Helms and co-workers found encephalopathic features in patients with severe covid-19 or SARS -Cov -2 [65]. The neurological features included, confusion, cognitive difficulties, poorly organised movement in response to command. Although it is too early at present to predict and also the situation is evolving, nevertheless, this sinister virus appears to have the potential to manifest some permanent long-term neurological defects.
Intriguingly, α-synuclein has also been suggested to operate as a natural antimicrobial peptide (AMP) at the front line of defence against invading pathogens (a component of the innate immune system). So, perhaps in the event of an attack by a pathogen, the α-synuclein comes to the defence initially, however it may undergo pathophysiological changes in the disease process (and in patients where it is overexpressed) resulting in its oligomerization, misfolding and subsequent accumulation.
The oligomerised form of α-synuclein can elicit neuronal destruction via membrane disruption, disruption of function of vital cellular organelles such as mitochondria and impairment of protein degradation. Thus, the aggregates of rogue α-synuclein (that is LB) may be responsible for the reduction in activity of mitochondrial complex I reported in SN in PD. These oligomerised α-synuclein may compromise the protein degradative mechanisms, thereby resulting in their accumulation. The ubiquitin-proteasome system and chaperone-mediated autophagy that are responsible for protein degradation may be overwhelmed by the build-up of the misfolded α-synuclein (Figure 2). It is also possible that they may not be fully operational under pathological conditions [23]. Subsequently, α-synuclein pathology propagates to neighbouring cells in a ‘’prion-like’’ fashion.
There are an array of factors which may contribute to the misfolding of α-synuclein, including familial PD related missense point mutation of α-synuclein gene, and some of them may affect the micro-environment for instance: a decrease in pH, an increase in temperature, amphipathic molecules (such as herbicides/pesticides), molecular crowding, polyamines, proteoglycans, chaotropic agents and metal ions [66]. Metal ions neutralise charge repulsion, which favours α-synuclein to a more condensed and aggregate-prone structure. It appears that the ability for metal ions to boost α-synuclein fibril formation is related to the ion charge density. Di- and trivalent metal ions such as aluminium, Iron (III), Copper (II) have been reported to be markedly efficient in this aspect [23].
Fascinatingly α-synuclein is able to operate as cellular ferrireductase. This enzyme reduces iron (III) to iron (II) using copper as a co-factor. In PD, a marked elevation in iron (III) and total iron has been reported in the SN [67]. Perhaps the α-synuclein/ferrireductase (Figure 2), is malfunctioning due to pathology of the disease or some genetic abnormality or endogenous/exo-toxin, this may result in disturbing the physiological iron homeostasis resulting in the deposition of iron [26]. The raised iron (III) content in SN may further exacerbate the neuronal destruction via augmenting of α-synuclein misfolding and accumulation, neuronal death via triggering cytotoxic processes such as oxidative stress.
Conclusion
There is compelling evidence supporting the involvement of the misfolded α-synuclein aggregates in the aetiology and pathogenesis of PD. There are number of mechanisms related to α-synuclein related neurotoxicity including toxic gain/loss of function, mechanical destruction of cellular processes/compartments. It is also possible that there are more than one-type of α-synuclein toxic aggregate, which may in turn employ different routes to induce neuronal death. These findings clearly demonstrate the malevolent actions of α-synuclein. It is therefore key to elucidate factors and mechanism(s) underlying the misfolding and accumulation of α-synuclein as this would provide an insight to the pathogenesis of the disease and thus effective treatment to halt its progression. However, it is unlikely that only a dysfunctional and threatening α-synuclein operates independently, it is rather more likely that other features such as genetic components, environmental factors, inflammatory agents are likely to make up this grand neurodegeneration orchestra. Neuro-inflammation observed in the nigral neurons may represent an epiphenomenon to the destruction of the dopaminergic cell loss in the SN in the early stages of the disease. However, it may subsequently contribute the nigral neuron fatalities.
References
2. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988 Aug 1;38(8):1285-91.
3. Stokholm MG, Iranzo A, Østergaard K, Serradell M, Otto M, Svendsen KB, et al. Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: a case-control study. The Lancet Neurology. 2017 Oct 1;16(10):789-96.
4. Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nature Reviews Neurology. 2010 Apr;6(4):193-201.
5. Chen H, Jacobs E, Schwarzschild MA, McCullough ML, Calle EE, Thun MJ, et al. Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 2005 Dec;58(6):963-7.
6. Gao X, Chen H, Schwarzschild MA, Ascherio A. Use of ibuprofen and risk of Parkinson disease. Neurology. 2011 Mar 8;76(10):863-9.
7. Durrenberger PF, Fernando FS, Kashefi SN, Bonnert TP, Seilhean D, Nait-Oumesmar B, et al. Common mechanisms in neurodegeneration and neuroinflammation: a BrainNet Europe gene expression microarray study. Journal of Neural Transmission. 2015 Jul 1;122(7):1055-68.
8. Lucin KM, Wyss-Coray T. Immune activation in brain aging and neurodegeneration: too much or too little?. Neuron. 2009 Oct 15;64(1):110-22.
9. Cherry JD, Olschowka JA, O’Banion MK. Neuroinflammation and M2 microglia: the good, the bad, and the inflamed. Journal of Neuroinflammation. 2014 Dec 1;11(1):98.
10. Kauppinen TM, Higashi Y, Suh SW, Escartin C, Nagasawa K, Swanson RA. Zinc triggers microglial activation. Journal of Neuroscience. 2008 May 28;28(22):5827-35.
11. Mogi M, Harada M, Kondo T, Riederer P, Inagaki H, Minami M, et al. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factoralpha are elevated in the brain from parkinsonian patients. Neuroscience Letters. 1994;180(2):147-50.
12. Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-a (TNF-a) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neuroscience letters. 1994 Jan 3;165(1-2):208-10.
13. Mogi M, Harada M, Kondo T, Narabayashi H, Riederer P, Nagatsu T. Transforming growth factor-ß1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neuroscience letters. 1995 Jun 30;193(2):129-32.
14. Mogi M, Harada M, Kondo T, Riederer P, Nagatsu T. Brain ß 2-microglobulin levels are elevated in the striatum in Parkinson’s diseaselevels are elevated in the striatum in Parkinson’s disease. Journal of Neural Transmission- Parkinson’s Disease and Dementia Section. 1995 Feb 1;9(1):87-92.
15. Mogi M, Harada M, Kondo T, Mizuno Y, Narabayashi H, Riederer P, et al. The soluble form of Fas molecule is elevated in parkinsonian brain tissues. Neuroscience Letters. 1996 Dec 20;220(3):195-8.
16. Mogi M, Harada M, Kondo T, Mizuno Y, Narabayashi H, Riederer P, et al. bcl-2 protein is increased in the brain from parkinsonian patients. Neuroscience Letters. 1996 Sep 6;215(2):137-9.
17. Jiang T, Yu JT, Tan L. Novel disease-modifying therapies for Alzheimer’s disease. Journal of Alzheimer’s disease. 2012 Jan 1;31(3):475-92.
18. Langston JW, Forno LS, Tetrud J, Reeves AG, Kaplan JA, Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4- phenyl-1, 2, 3, 6-tetrahydropyridine exposure. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1999 Oct;46(4):598-605.
19. Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. European Journal of Neuroscience. 2002 Mar;15(6):991-8.
20. Liu Y, Qin L, Li G, Zhang W, An L, Liu B, et al. Dextromethorphan protects dopaminergic neurons against inflammation-mediated degeneration through inhibition of microglial activation. Journal of Pharmacology and Experimental Therapeutics. 2003 Apr 1;305(1):212-8.
21. Iacono D, Geraci-Erck M, Rabin ML, Adler CH, Serrano G, Beach TG, et al. Parkinson disease and incidental-Lewy body disease: just a question of time?. Neurology. 2015 Nov 10;85(19):1670-9.
22. Breydo L, Wu JW, Uversky VN. a-Synuclein misfolding and Parkinson’s disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2012 Feb 1;1822(2):261-85.
23. Riederer P, Berg D, Casadei N, Cheng F, Classen J, Dresel C, et al. a-Synuclein in Parkinson’s disease: causal or bystander?. Journal of Neural Transmission. 2019 Jul 1;126(7):815-40.
24. Watson JB, Hatami A, David H, Masliah E, Roberts K, Evans CE, Levine MS. Alterations in corticostriatal synaptic plasticity in mice overexpressing human a-synuclein. Neuroscience. 2009 Mar 17;159(2):501-13.
25. Gibb WR, Lees A. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. Journal of Neurology, Neurosurgery & Psychiatry. 1988 Jun 1;51(6):745-52.
26. Sian-Hulsmann J, Riederer P. The Role of Alpha- Synuclein as Ferrireductase in Neurodegeneration Associated With Parkinson’s Disease. Journal of Neural Transmission. 2020. 127(5):749-754.
27. Braak H, Del Tredici K, Rüb U, De Vos RA, Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging. 2003 Mar 1;24(2):197-211.
28. Koros C, Simitsi A, Stefanis L. Genetics of Parkinson’s disease: genotype–phenotype correlations. International Review of Neurobiology. 2017 Jan 1;132:197-231.
29. Singleton AB , Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, et al. [alpha]-Synuclein Locus Triplication Causes Parkinson’s Disease. Science. 2003 Oct 31;302(5646):841-2.
30. Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular a-synuclein aggregates in microglia. Biochemical and Biophysical Research Communications. 2008 Aug 1;372(3):423-8.
31. Tagliafierro L, Chiba-Falek O. Up-regulation of SNCA gene expression: implications to synucleinopathies. Neurogenetics. 2016 Jul 1;17(3):145-57.
32. Uversky VN, Yamin G, Munishkina LA, Karymov MA, Millett IS, Doniach S, et al. Effects of nitration on the structure and aggregation of a-synuclein. Molecular Brain Research. 2005 Mar 24;134(1):84-102.
33. Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize a-synuclein peptides. Nature. 2017 Jun;546(7660):656-61.
34. Brochard V, Combadiere B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of Clinical Investigation. 2019;119:182-92.
35. Jellinger KA. Neuropathology of sporadic Parkinson’s disease: evaluation and changes of concepts. Movement Disorders. 2012 Jan;27(1):8-30.
36. Nielsen HH, Qiu J, Friis S, Wermuth L, Ritz B. Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. European Journal of Neurology. 2012 Jun;19(6):864-9.
37. Choi JG, Kim N, Ju IG, Eo H, Lim SM, Jang SE, et al. Oral administration of Proteus mirabilis damages dopaminergic neurons and motor functions in mice. Scientific Reports. 2018 Jan 19;8:1275.
38. Foley PB. Encephalitis lethargica and influenza. I. The role of the influenza virus in the influenza pandemic of 1918/1919. Journal of Neural Transmission. 2009 Feb 1;116(2):143-50.
39. Lutters B, Foley P, Koehler PJ. The centennial lesson of encephalitis lethargica. Neurology. 2018 Mar 20;90(12):563-7.
40. Elizan TS, Casals J. The viral hypothesis in parkinsonism. Journal of Neural Transmission. Supplementum. 1983 Jan 1;19:75-88.
41. Takahashi M, Yamada T. A possible role of influenza A virus infection for Parkinson’s disease. Advances in Neurology. 2001;86:91-104.
42. Hawkes CH, Del Tredici K, Braak H. Parkinson’s disease: a dual-hit hypothesis. Neuropathology and Applied Neurobiology. 2007 Dec;33(6):599-614.
43. Marttila RJ, Halonen P, Rinne UK. Influenza virus antibodies in parkinsonism: Comparison of postencephalic and idiopathic Parkinson patients and matched controls. Archives of Neurology. 1977 Feb 1;34(2):99-100.
44. Elizan TS, Madden DL, Noble GR, Herrmann KL, Gardner J, Schwartz J, et al. Viral antibodies in serum and CSF of Parkinsonian patients and controls. Archives of neurology. 1979 Sep 1;36(9):529-34.
45. Jellinger KA. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. Journal of Neuropathology & Experimental Neurology. 2001 Nov;60(11):1121-2.
46. McCall S, Henry JM, Reid AH, Taubenberger JK. Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. Journal of Neuropathology & Experimental Neurology. 2001 Jul 1;60(7):696-704.
47. Schwartz J, Elizan TS. Search for viral particles and virus-specific products in idiopathic Parkinson disease brain material. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1979 Sep;6(3):261-3.
48. Gamboa ET, Wolf A, Yahr MD, Harter DH, Duffy PE, Barden H, et al. Influenza virus antigen in postencephalitic parkinsonism brain: detection by immunofluorescence. Archives of Neurology. 1974 Oct 1;31(4):228-32.
49. Mihara M, Utsugisawa K, Konno S, Tohgi H. Isolated lesions limited to the bilateral substantia nigra on MRI associated with influenza A infection. European Neurology. 2001 May 1;45(4):290.
50. Arbour N, Day R, Newcombe J, Talbot PJ. Neuroinvasion by human respiratory coronaviruses. J Virol. 2000 Oct;74(19):8913-21.
51. Rohn TT, Catlin LW. Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PloS one. 2011 May 31;6(5):e20495.
52. Harris MA, Tsui JK, Marion SA, Shen H, Teschke K. Association of Parkinson’s disease with infections and occupational exposure to possible vectors. Movement Disorders. 2012 Aug;27(9):1111-7.
53. Attanasio R. Environmental Toxins and Damage to the Immune System: Transgenerational Effects. 2019.
54. Jellinger KA. Neuropathology and pathogenesis of extrapyramidal movement disorders: a critical update—I. Hypokinetic-rigid movement disorders. Journal of Neural Transmission. 2019 Aug 1;126(8):933-95.
55. Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, Beckham JD. Alphasynuclein expression restricts RNA viral infections in the brain. Journal of Virology. 2016 Mar 15;90(6):2767-82.
56. Khanlou N, Moore DJ, Chana G, Cherner M, Lazzaretto D, Dawes S, et al. Increased frequency of a-synuclein in the substantia nigra in human immunodeficiency virus infection. Journal of Neurovirology. 2009 Mar 1;15(2):131- 8.
57. Mackiewicz MM, Overk C, Achim CL, Masliah E. Pathogenesis of age-related HIV neurodegeneration. Journal of Neurovirology. 2019 Oct 1;25(5):622-33.
58. Itoh K, Mehraein P, Weis S. Neuronal damage of the substantia nigra in HIV-1 infected brains. Acta Neuropathologica. 2000 Mar 1;99(4):376-84.
59. Koutsilieri E, Götz ME, Sopper S, Sauer U, Demuth M, ter Meulen V, et al. Regulation of glutathione and cell toxicity following exposure to neurotropic substances and human immunodeficiency virus-1 in vitro. Journal of Neurovirology. 1997 Jan 1;3(5):342-9
60. Scheller C, Sopper S, Jassoy C, ter Meulen V, Riederer P, Koutsilieri E. Dopamine activates HIV in chronically infected T lymphoblasts. Journal of Neural Transmission. 2000 Dec 1;107(12):1483-9.
61. Koutsilieri E, ter Meulen V, Riederer P. Neurotransmission in HIV associated dementia: a short review. J Neural Transm. (2001). 108(6):767-75.
62. Meisner F, Scheller C, Kneitz S, Sopper S, Neuen- Jacob E, Riederer P, et al. Memantine Upregulates BDNF and Prevents Dopamine Deficits in SIV-infected Macaques: A Novel Pharmacological Action of Memantine. Neuropsychopharmacology. 2008 Aug;33(9):2228-36.
63. Tan SV, Guiloff RJ. Hypothesis on the pathogenesis of vacuolar myelopathy, dementia, and peripheral neuropathy in AIDS. Journal of Neurology, Neurosurgery & Psychiatry. 1998 Jul 1;65(1):23-8.
64. Horn A, Scheller C, du Plessis S, Arendt G, Nolting T, Joska J, et al. Increases in CSF dopamine in HIV patients are due to the dopamine transporter 10/10-repeat allele which is more frequent in HIV-infected individuals. Jou
65. Helms J, Kremer S, Merdji H, Clere-Jehl R, Schenck M, Kummerlen C, et al. Neurologic Features in Severe SARS-CoV-2 Infection. New England Journal of Medicine. 2020 June 4;382:2268-70.
66. Candelise N, Schmitz M, Thüne K, Cramm M, Rabano A, Zafar S, et al. Effect of the micro-environment on a-synuclein conversion and implication in seeded conversion assays. Translational Neurodegeneration. 2020 Dec;9(1):1-6.
67. Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. Journal of Neural Transmission. 1988 Oct 1;74(3):199-205.