Commentary
N6-fufuryladenine (N6FFA), or kinetin, has a long history as a plant cytokine with practical applications in agriculture. This adenosine analog is now commonplace in natural product small molecule chemical screening libraries, and as such has been discovered as active in mammalian disease pathways that include Parkinson’s disease, Huntington’s disease (HD) and Familial Dysautonomia. We provide a perspective on our data relative to HD and recent discoveries of genetic modifiers of this disease predominantly related to DNA damage repair. We outline the importance of nucleotide salvage and the presence of this adenosine analog in human samples and efficacy in models of human disease, with mechanisms that are empowered by chemical studies using N6FFA as a nucleic acid crosslinking agent.
In July 2018, we published a report on the discovery of a lead compound, N6-furfuryladenine (N6FFA or kinetin), from a library of bioactive compounds, that restored the hypo-phosphorylation of the mutant huntingtin protein within the N17 domain [1]. N17 is a 17 amino acid domain within huntingtin adjacent to the CAG DNA expanded polyglutamine tract that appears to be a master switch regulator of huntingtin localization from the endoplasmic reticulum and vesicles to the nucleus and DNA damage sites [2,3]. N17 can be modified at position serine 13 or 16 by casein kinase 2 (CK2) [4]. The screen was conducted in an unbiased manner, with robotic microscopy data acquisition and subsequent non-supervised machine sorting with Principal Component Analysis [5], then secondary and tertiary assays to directly measure the effect of the compound on the restoration of a serine phosphorylation in huntingtin N17. This compound was then shown in mouse cortical neurons and a HD mouse model to revert some disease phenotypes, and lower the load of mutant huntingtin aggregates in the HD mouse brain. This lead compound activity was rare in that it restored a signaling pathway seen defective in disease.
Early days of understanding how this compound was mechanistically acting in our system was confounded by 40 years of publications that only defined N6FFA as a plant growth cytokine, or mammalian applications in dermatology as an applied topical agent [6]. In human cell studies, N6FFA was typically defined as a plant-based compound, but it was only after a thorough collation of all sources of data on this compound in dermatology, industry, and cancer research, did we start to understand that N6FFA is a universal by-product of DNA modification by the Fenton reaction, which could be triggered by simple genotoxic stresses like heat or reactive oxygen [7]. One critical study was the discovery of N6FFA in human urine, and the elevation of levels in human cancer, demonstrating this compound was produced in humans [8].
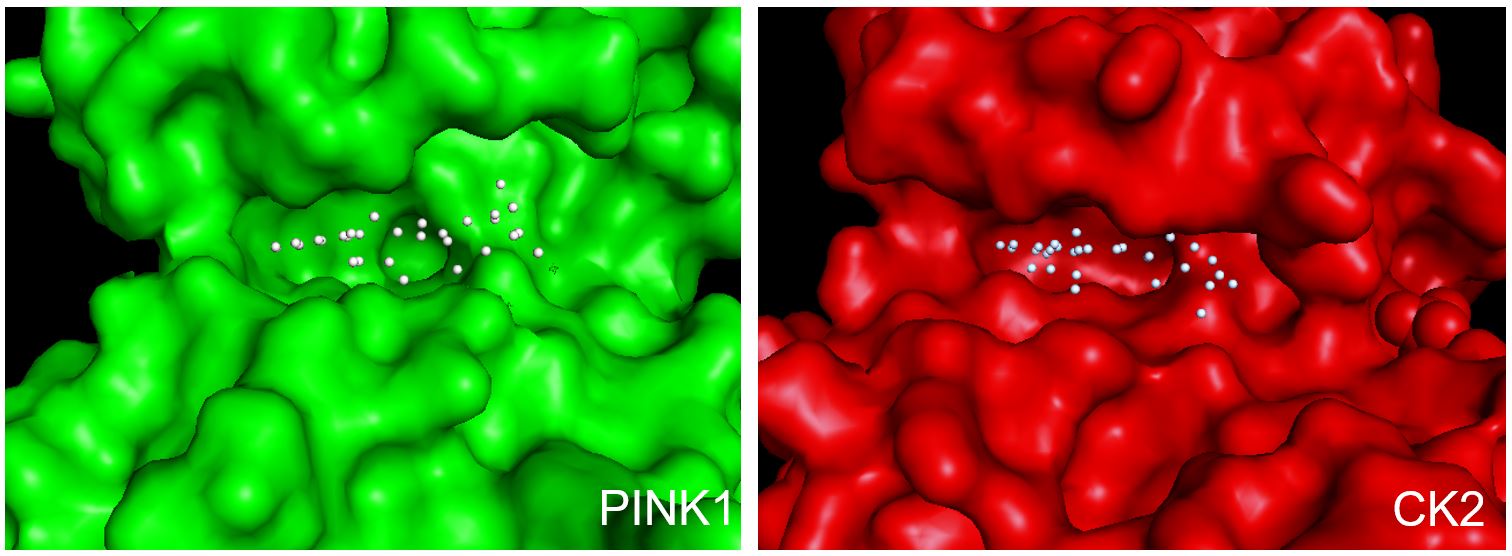
From high-content screening by others, N6FFA was discovered in 2013 to restore the activity of the mutant PINK1 kinase that is causative to a rare genetic Parkinson’s disease [9]. PINK1 has been found to localize to the outer membrane of mitochondria, and is detected in the cytosol where it is thought to sense mitochondrial damage and trigger mitophagy via Parkin signaling. Similar to HD, in Parkinson’s disease there appeared to be a defective kinase signaling pathway. This type of drug target has not been pursued by industry, as almost all of the kinase targets pursued involve kinase inhibition as a disease therapeutic. One critical mechanism discovered by Hertz et al., was that N6FFA was being salvaged from an adenosine analog to a triphosphate analog, kinetin triphosphate, or KTP. This salvaging was being done by adenine phosphoribosyltransferase, or APRT. Thus, Hertz defined KTP as a “neo-substrate”, an alternative to ATP or GTP as a phosphate donor that can still be catalytically used by the mutant PINK1. What was then intriguing was that two independent groups focused on two different age-onset genetic neurodegenerative diseases commonly discovered a similar lead compound that in both cases restored a defective signaling pathway, yet through two distinct kinases. What is evident from the structures of the catalytic domains of CK2 and PINK1 is that they both have an unusually large void around the ATP pocket to accommodate the furfuryl sidegroup in N6FFA and potentially other N6-modified adenosine analogs (Figure 1).
Parallel projects within the lab by cell biology were leading us to conclude huntingtin had a role in DNA damage repair as a scaffold, and that importantly, that this was signaled by CK2 [10]. The relevance to this in human disease became paramount as the result of a series of very large genetic modifiers of disease studies came to the conclusion that DNA repair factors were major modifiers of HD age at onset [11-13]. It was at this point we realized that N6FFA as a cytokine, and huntingtin as a DNA repair protein, were in fact interrelated. This led the hypothesis of a biochemical signaling feedback loop, in which huntingtin activity was responsible for the correction of N6FFA adducts in damaged DNA, which netted free N6FFA, which was then salvaged to KTP, and used by CK2 to modify huntingtin to feedback to more DNA repair. As N6FFA adducts are repaired, the signal is naturally dampened as KTP is used up. In HD, the lack of N6FFA repair led to a lack of downstream KTP, hence resulting in a loss of function of mutant huntingtin and a defective signaling pathway. By adding N6FFA in trans, allowing for salvaging to KTP, we then pushed forward this signaling reaction and hence showed benefit in mouse models.
Revisiting the Effects of N6FFA in Humans
N6FFA was discovered in dermatology research to provide benefit on epithelial cell health [14]. While a mechanism of anti-oxidant was proposed, N6FFA itself does not have any anti-oxidant chemical properties, but does signal an antioxidant response [15], likely tied to DNA damage events. N6FFA is now an additive in cosmetic topical skin creams, and benefits to skin are likely a combination of activation of antioxidant response pathways as well as activating DNA damage repair, which is a major stress event in skin epithelial cells. This has led to the term, “cosmeceutical” [16].
N6FFA was also discovered in a screen to correct a mRNA splicing defect seen in familial dysautonomia (FD), a pediatric neurological disorder caused by a splice defect in ELP1 gene [17]. This results in a defective kappa B kinase complex-associated protein (IKAP) protein. In mouse models of FD, dosing from birth could restore proper ELP1 RNA splicing, increase IKAP protein levels, and reduce the phenotype of proprioceptive sensory loss [18]. Human clinical trials of N6FFA were initiated for FD [17], but as a sparingly soluble compound, pharmacological preparations to achieve dose levels scaled from mice to humans have a challenge with oral bioavailability. At this point, the exact molecular mechanism of how N6FFA leads to splicing correction is unknown, but it is intriguing that a role of huntingtin in transcription-coupled DNA repair has been recently discovered [19]. There may be an effect of correction of ELP1 mRNA mis-splicing during transcription-coupled repair via enhanced CK2 activity by the presence of KTP.
Could the Huntington’s-Parkinson’s N6FFA Connection be TP53?
Upstream of the huntingtin/CK2/KTP feedback loop, CK2 is well defined as a signaling kinase in DNA damage response [20]. One of the targets of CK2 modification is the tumor suppressor protein, TP53[21]. Upon modification of serine 386 by CK2, TP53 is prompted into its tetrameric transcriptionally active state [21]. Active TP53 can increase transcription and production of huntingtin via a TP53 response element in the HTT promoter [22]. Thus, we have a higher level of sensing and regulation, as TP53 is historically defined as a genomic integrity sentinel, constantly shuttling between nucleus and cytoplasm to monitor DNA integrity. As DNA damage is detected by TP53, the result is higher transcription and protein levels of huntingtin. The chronic high expression levels of mutant huntingtin in HD brain suggests TP53 may be sensing increased DNA damage and one result of that is to increase huntingtin levels, but mutant huntingtin cannot correct DNA damage. At a threshold of DNA damage detection, TP53 may then be signaling neuronal death via apoptosis to prevent tumorigenesis.
When an HD model mouse was dosed with N6FFA, we observed a surprising drop in the levels of detectable protein inclusions, or insoluble aggregates, of mutant huntingtin in the brain in a dose-response manner. We hypothesize that priming the signaling with exogenous N6FFA, hence KTP, led to reduced DNA damage and hence a dampening of TP53 activation of huntingtin. The analogy is a sink overflowing with water, while a classic approach of therapeutics is to reduce huntingtin levels (the “water”), this does not address the tap (TP53) continuing to pour water. By reducing damage, hence CK2 signaling, hence TP53 activity, N6FFA/KTP could shut off the “tap”.
Another target of active TP53 is PINK1, but in that case, TP53 activity represses PINK1 activity and hence suppresses mitophagy [23]. There is thus inter-regulation between CK2 and PINK1, where KTP can be used to decrease PINK1 transcription via CK2, but also be used to increase PINK1 kinase activity on PINK1 substrates. At this point, the net beneficial pathway for neurodegenerative disease is not known. CK2 modification of TP53 leads to a bifurcated response, increasing huntingtin and active phospho-huntingtin, as well as down-regulation of mitophagy, which may benefit a stressed neuron at a time of severe energy deficit, utilizing inefficient mitochondria as opposed to the genesis of new mitochondria, which is energy intensive. While we focused on the use of KTP to modify huntingtin, we also used KTP across a panel of known CK2 sites in proteins to conclude CK2 can use KTP on any classic substrate, including many proteins in DNA damage repair [1].
The Chemistry of N6FFA DNA Adducts, and the Importance of FAN1 in HD
The major genetic modifier of HD identified by Genome- Wide Association Studies of over 12,000 HD patients was the Fanconi Anemia DNA repair factor, FAN1 [24]. Follow up studies in HD patients have shown higher FAN1 protein levels to correlate with later age at onset, and less somatic expansion of the CAG DNA repeat in HTT [25]. FAN1 is well defined as a DNA repair factor endonuclease that acts to correct DNA interstrand crosslinks (ICL). While FAN1 levels correlate to the stability of CAG tract in HTT, neither the exact mechanism, nor the mechanistic consequences of CAG hyper-expansion are known.
N6FFA in DNA has been under development as a chemical tool to promote cross-linking with proteins and nucleic acid, both DNA and RNA, via furan reactivity [26]. By the use of the furan side group in N6-furfuryladenine, inter and intra cross links can be created between nucleic acids and proteins [27], and this chemistry is promoted by reactive oxygen levels [28]. Thus, the combination of increased ageing reactive oxygen stress and the failure to correct N6FFA adducts may promote DNA cross-linking in HD, making the activity of FAN1 more important to correct these ICLs. Future work on quantifying trapped N6FFA in HD neuronal DNA as well as the mechanistic implications of ICLs leading to CAG DNA instability could give us very precise mechanistic insights into the beneficial role of FAN1 activity in HD and hence new practical therapeutic targets. One area that requires further study is the presence of N6FFA DNA adducts and any effect on CAG DNA somatic instability in the huntingtin gene, as current mechanistic models of DNA instability assume unmodified DNA. An appealing concept is that typical reactive oxygen stress increases with human ageing may trigger more N6FFA adducts and hence lead to more instability of the CAG tract in the huntingtin gene, thus amplifying the effect of the mutation in the huntingtin mRNA or protein. This could partially explain the ageonset nature of HD. This suggests huntingtin activity in DNA repair and dysfunction of mutant huntingtin may be contributing to the somatic expansion of its own gene. Indeed, one intriguing study indicates the CAG expansion in another CAG repeat disease gene, spinocerebellar ataxia type 2 (SCA2), can be affected by huntingtin levels [29].
Currently, an effort to derive a chemical series of N6FFA derivatives with improved potency and pharmacokinetics is underway at Mitokinin Inc., a biotech company located in San Francisco, California. Mitokinin has active programs in Parkinson’s disease and Huntington’s disease.
Acknowledgements
The Truant laboratory is supported by the Krembil Foundation, Toronto, Canada.
Declaration of Conflicts
RT is a member of the Scientific Advisory Board and shareholder, Mitokinin Inc.
References
2. Maiuri T, Bowie LE, Truant R. DNA Repair Signaling of Huntingtin: The Next Link Between Late-Onset Neurodegenerative Disease and Oxidative DNA Damage. DNA Cell Biol. 2019;38: 1-6.
3. Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16: 2600-2615.
4. Atwal RS, Desmond CR, Caron N, Maiuri T, Xia J, Sipione S, et al. Kinase inhibitors modulate huntingtin cell localization and toxicity. Nat Chem Biol. 2011;7: 453-460.
5. Rajaram S, Pavie B, Wu LF, Altschuler SJ. PhenoRipper: software for rapidly profiling microscopy images. Nat Methods. 2012;9: 635-637.
6. Hönig M, Plíhalová L, Spíchal L, Grúz J, Kadlecová A, Voller J, et al. New cytokinin derivatives possess UVA and UVB photoprotective effect on human skin cells and prevent oxidative stress. Eur J Med Chem. 2018;150: 946- 957.
7. Olsen A, Siboska GE, Clark BF, Rattan SI. N(6)- Furfuryladenine, kinetin, protects against Fenton reactionmediated oxidative damage to DNA. Biochem Biophys Res Commun. 1999;265: 499-502.
8. Barciszewski J, Mielcarek M, Stobiecki M, Siboska G, Clark BF. Identification of 6-furfuryladenine (kinetin) in human urine. Biochem Biophys Res Commun. 2000;279: 69-73.
9. Hertz NT, Berthet A, Sos ML, Thorn KS, Burlingame AL, Nakamura K, et al. A Neo-Substrate that Amplifies Catalytic Activity of Parkinson's-Disease-Related Kinase PINK1. Cell. 2013;154: 737-747.
10. Maiuri T, Mocle AJ, Hung CL, Xia J, van Roon-Mom WMC, Truant R. Huntingtin is a scaffolding protein in the ATM oxidative DNA damage response complex. Hum Mol Genet. 2017;26: 395-406.
11. Correia K, Harold D, Kim K-H, Holmans P, Jones L, Orth M, et al. The Genetic Modifiers of Motor OnsetAge (GeM MOA) Website: Genome-wide Association Analysis for Genetic Modifiers of Huntington’s Disease. J Huntingtons Dis. 2015;4: 279-284.
12. Moss DJH, Pardiñas AF, Langbehn D, Lo K, Leavitt BR, Roos R, et al. Identification of genetic variants associated with Huntington’s disease progression: a genome-wide association study. Lancet Neurol. 2017.
13. Genetic Modifiers of Huntington's Disease (GeM-HD) Consortium. Identification of Genetic Factors that Modify Clinical Onset of Huntington's Disease. Cell. 2015;162: 516-526.
14. Kowalska E. Influence of kinetin (6-furfurylo-aminopurine) on human fibroblasts in the cell culture. Folia Morphol. 1992;51:109-118.
15. Jablonska-Trypuc A, Matejczyk M, Czerpak R. N6- benzyladenine and kinetin influence antioxidative stress parameters in human skin fibroblasts. Mol Cell Biochem. 2016;413: 97-107.
16. Kligman D. Cosmeceuticals. Dermatol Clin. 2000;18: 609-615.
17. Axelrod FB, Liebes L, Gold-Von Simson G, Mendoza S, Mull J, Leyne M, et al. Kinetin improves IKBKAP mRNA splicing in patients with familial dysautonomia. Pediatr Res. 2011;70: 480-483.
18. Morini E, Gao D, Montgomery CM, Salani M, Mazzasette C, Krussig TA, et al. ELP1 Splicing Correction Reverses Proprioceptive Sensory Loss in Familial Dysautonomia. Am J Hum Genet. 2019;104: 638-650.
19. Gao R, Chakraborty A, Geater C, Pradhan S, Gordon KL, Snowden J, et al. Mutant huntingtin impairs PNKP and ATXN3, disrupting DNA repair and transcription. Elife. 2019;8.
20. Montenarh M. Protein kinase CK2 in DNA damage and repair. Transl Cancer Res. 2016;5: 49-63.
21. McKendrick L, Milne D, Meek D. Protein kinase CK2- dependent regulation of p53 function: evidence that the phosphorylation status of the serine 386 (CK2) site of p53 is constitutive and stable. Mol Cell Biochem. 1999;191: 187-199.
22. Feng Z, Jin S, Zupnick A, Hoh J, de Stanchina E, Lowe S, et al. p53 tumor suppressor protein regulates the levels of huntingtin gene expression. Oncogene. 2006;25: 1-7.
23. Checler F, Goiran T, Alves da Costa C. Nuclear TP53: An unraveled function as transcriptional repressor of PINK1. Autophagy. 2018. pp. 1099-1101.
24. Kim K-H, Hong EP, Shin JW, Chao MJ, Loupe J, Gillis T, et al. Genetic and Functional Analyses Point to FAN1 as the Source of Multiple Huntington Disease Modifier Effects. Am J Hum Genet. 2020;107: 96-110.
25. Goold R, Flower M, Moss DH, Medway C, Wood- Kaczmar A, Andre R, et al. FAN1 modifies Huntington’s disease progression by stabilizing the expanded HTT CAG repeat. Hum Mol Genet. 2019;28: 650-661.
26. Carrette LLG, Gyssels E, De Laet N, Madder A. Furan oxidation based cross-linking: a new approach for the study and targeting of nucleic acid and protein interactions. Chem Commun. 2016;52: 1539-1554.
27. Carrette LLG, Gyssels E, Madder A. DNA interstrand cross-link formation using furan as a masked reactive aldehyde. Curr Protoc Nucleic Acid Chem. 2013;54: 5.12.1- 5.12.16.
28. Antonatou E, Hoogewijs K, Kalaitzakis D, Baudot A, Vassilikogiannakis G, Madder A. Singlet Oxygen-Induced Furan Oxidation for Site-Specific and Chemoselective Peptide Ligation. Chemistry. 2016;22: 8457-8461.
29. Coffey SR, Andrew M, Ging H, Hamilton J, Flower M, Kovalenko M, et al. Huntingtin lowering reduces somatic instability at CAG-expanded loci. bioRxiv. 2020. p. 2020.07.23.218347.