Commentary
Since the development of Ceredase® and Cerezyme® for the treatment of Gaucher disease in the early 1990s, treatment of lysosomal storage disorders via enzyme replacement therapy (ERT) has provided life-changing benefit to patients and their families. Treating the neurological symptoms of these rare diseases, however, remains a significant unmet medical need. Here, we focus, as commentary on the recent publication by Grover et al, on the mucopolysaccharidosis (MPS) family of lysosomal storage diseases, in which one of enzymes required to break down the substrate of these enzymes, glycosaminoglycans (GAGs), are deficient [1]. In the absence of the enzyme, GAGs accumulate in systemic and central nervous system (CNS) tissues, leading to significant morbidity and mortality.
The MPSs are, in general, monogenic diseases resulting from the complete deficiency or substantially reduced function of one of eleven lysosomal enzymes involved in GAG metabolism. They are autosomal recessive disorders, with the exception of MPS II, which is X-linked recessive. Today, these diseases are generally diagnosed via an enzyme activity assay or genetic sequencing. Elevated GAG levels in fluids, primarily urine, may also serve as preliminary evidence of disease.
Grover et al. [1], present translational support for the development of BMN 250, a treatment of Sanfilippo Type B, also known as MPS IIIB, currently in clinical study. BMN 250 is administered directly into the cerebrospinal fluid through a catheter inserted into a lateral ventricle, known as intracerebroventricular (ICV) administration. The preclinical studies presented by Grover et al. [1] present a framework for translational research to address this unmet medical need of neurological symptoms of MPS diseases.
The blood-brain barrier (BBB) is evolutionarily adapted to protect the brain parenchyma from foreign molecules entering through the intravascular space, including enzymes administered peripherally as enzyme replacement therapy. The BBB is composed of capillary endothelial cells, astrocyte end-feet, capillary basement membrane, and pericytes within the basement membrane. Therefore, intravenously-administered enzymes that treat the peripheral manifestations of disease do not treat the neurological manifestations of MPSs. Numerous approaches to treat the CNS manifestations of disease have been studied in clinical trials or are planned to be studied and are highlighted in Table 1. In Table 2, a summary of clinical programs for the treatment of the CNS manifestations of MPS diseases that focus on restoring the specific enzyme in defect (i.e., not including stem cell transplant or supportive programs such as nutrients or anti-inflammatories) and in which dose can be fine-tuned (i.e., not including ex vivo gene therapy) as of April 2020 are listed. These include direct approaches to bypass the BBB, such as the ICV administration studied in Grover et al. study [1], or protein engineering approaches with intravenous administration. Some programs listed in Table 2 have been terminated due to lack of demonstration of efficacy but are included for completeness. The multiplicity of approaches here, in contrast to systemic therapies for MPS diseases, is evidence that the “best” approach is yet to be determined.
| Approach | Advantages | Limitations |
|---|---|---|
| Direct-to-CSF ERT |
|
|
| Direct-to-CSF gene therapy |
|
|
| Intraparenchymal gene therapy |
|
|
| IV-administered, protein-engineered or protein- optimized ERT to cross the BBB |
|
|
| IV-administered, CNS-targeted gene therapy |
|
|
| IV-administered, liver-targeted gene therapy |
|
|
Table 1: Approaches to target the CNS for the treatment of MPS diseases.
| Disease | Therapeutic | Approach | clinicaltrials.gov Reference |
|---|---|---|---|
| MPS I | |||
| AGT-181 | Protein engineered ERT via insulin receptor-mediated uptake following IV administration | NCT03053089 | |
| JR-171 | Protein engineered ERT via transferrin receptor- mediated uptake following IV administration | NCT04227600 | |
| Laronidase | ERT via the IT-L route | NCT00852358 | |
| RGX-111 | Gene therapy targeting CNS tissue via the IC route | NCT03580083 | |
| SB-318 | Gene therapy targeting liver via the IV route | NCT02702115 | |
| MPS II | |||
| AGT-182 | Protein engineered ERT via insulin receptor-mediated uptake following IV administration | NCT02262338 | |
| DNL310 | Protein engineered ERT via transferrin receptor- mediated uptake following IV administration | NCT04251026 | |
| Idursulfase | ERT via the IT-L route | NCT00920647 | |
| JR-141 | Protein engineered ERT via transferrin receptor- mediated uptake following IV administration | NCT03568175 | |
| RGX-121 | Gene therapy targeting CNS tissue via the IC route | NCT03566043 | |
| SB-913 | Gene therapy targeting liver via the IV route | NCT03041324 | |
| MPS IIIA | |||
| ABO-102/scAAV9.U1a. hSGSH |
Gene therapy targeting CNS tissue via the IV route | NCT02716246 | |
| LYS-SAF302 | Gene therapy via direct administration into brain parenchyma | NCT03612869 | |
| rhNHS | ERT via the IT-L route | NCT01155778 | |
| SAF-301 | Gene therapy via direct administration into brain parenchyma | NCT01474343 | |
| SOBI0003 | Glycan-modified ERT | NCT03423186 | |
| MPS IIIB | |||
| AX 250 (formerly BMN 250) | ERT via the ICV route | NCT02754076 | |
| rAAV2/5-hNAGLU | Gene therapy via direct administration into brain parenchyma | NCT03300453 | |
| rAAV9.CMV.hNAGLU | Gene therapy targeting CNS tissue via the IV route | NCT03315182 | |
| SBC-103 | Glycan-modified ERT following IV administration (uptake via CI-MPR) |
NCT02324049 |
Results from clinicaltrials.gov search of terms: MPS + CNS, MPS + neurological, MPS + cognitive, MPS + CSF, MPS + cerebrospinal fluid, mucopolysaccharidosis + cerebrospinal fluid, mucopolysaccharidosis + cognitive, Sanfilippo. Not all programs are active. Stem cell transplant studies, supportive therapies such as nutrients or anti-inflammatories, and ex vivo gene therapy studies are not included. A single clinicaltrials.gov listing is provided for programs with multiple studies.
CI-MPR: Cation-Independent Mannose-6-Phosphase Receptor; ERT: Enzyme Replacement Therapy; IC: Intra- Cisternal; ICV: Intracerebroventricular; IT-L: Intrathecal-Lumbar; IV: Intravenous
Table 2: Experimental products in clinical trials for the treatment of neurological manifestations of MPS diseases.
The first direct approach is administration into the CSF, including via the intrathecal-lumbar, intrathecal cisternal, or intracerebroventricular (also known as intraventricular) routes (Figure 1). This approach is supported by the approvals of Brineura® (cerliponase alfa), an enzyme replacement therapy for the treatment of a form of Battens disease, late infantile neuronal ceroid lipofuscinosis type 2 (CLN2, resulting from deficient TPP1 enzyme), via the intraventricular route, and Spinraza® (nusinersen), an oligonucleotide for the treatment of spinal muscular atrophy via the intrathecal-lumbar route, in the non-ERT space. Direct-to-CSF approaches include both replacement of the enzyme itself or gene therapies that use CNS cell types as manufacturing sites for enzyme.
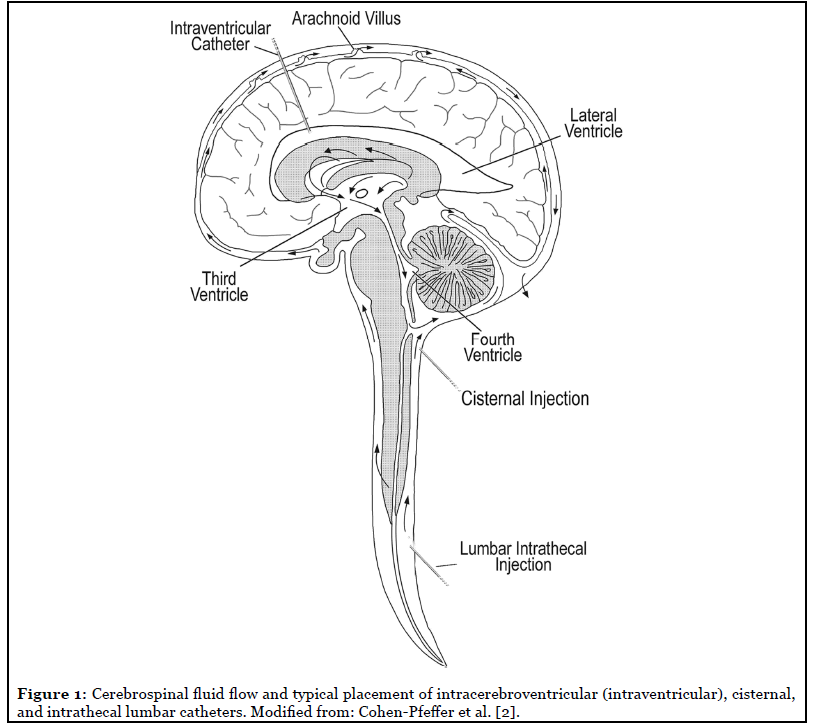
A comparatively more invasive approach is administration directly into the brain tissue, such as via intraparenchymal injection. This approach is generally only feasible for gene therapy, in which a single administration is hoped to be sufficient for life-long benefit, and it relies on an assumption of cross-correction between cells: that is, enzyme produced in a “gene therapy-infected” cell is released and able to be taken up into a nearby, “uninfected” cell.
With any of these approaches to bypass or cross the BBB, there are a few key sets of data that support drug development, as highlighted in Grover et al. study [1].
First, in the preclinical work supporting drug development, it is critical to demonstrate enzyme levels and/or GAG substrate reduction in cell types relevant to disease. That is, most generally, neurons, astrocytes, glia, and/or oligodendrocytes. Various approaches have been utilized towards this end, including immunohistochemistry [1,3] or florescence-activated cell sorting [4]. Importantly, as demonstrated by Grover et al. [1], intravenously administered enzyme may be trapped in the endothelial cells comprising the blood-brain barrier, such that enzyme does not reach its target cell types, and therefore it is critically important to ensure action in the target cell types during drug development for these intravenous, protein engineering approaches.
Similarly, regional drug biodistribution with the brain is often assessed in a large animal species, such as cynomolgus monkey, to ensure each brain region receives sufficient drug levels. For administration routes directly into the CSF, this may include assessments of both superficial and deep, relative to CSF flow, tissues [1,5].
A fundamental question in a clinical study of such a program centers of the value of enzyme and GAG levels in the CSF. In patient studies, CSF is the only CNS tissue able to be sampled while the patient is treated. From a pharmacokinetic, or drug concentration, perspective, drug is rapidly taken up into tissue following administration into the CSF, and the drug kinetics of CSF will not necessarily represent those in brain. For example, following ICV administration to cynomolgus monkeys, the half-life of cerliponase alfa in CSF was 2-4 hours, while the brain tissue half-life was generally 3-15 days across tissues (Brineura® summary basis of approval, fda.gov). The observed CSF half-life in patients was approximately 6-8 hours, similar to that observed in cynomolgus monkey (Brineura® summary basis of approval, fda.gov). Preclinical work should also prioritize understanding the relationship between CSF response and brain tissue response. Perfused brain homogenate from pharmacology studies using animal models of disease may be the only quantitatively representative assessment of whole brain, as neither IHC nor FACS are truly independently quantitative, and thus, while crude, brain homogenate may be used to correlate brain pharmacological response with that in the CSF [6]. As noted above, however, cell-specific distribution of enzyme or GAG response should also be assessed in order to differentiate between distribution or drug effect limited to the endothelial cells. Importantly, such CSF:brain GAG correlations in animals are generally created from tightly controlled animal studies, e.g., animals of the same genotype, approximate age, etc. In the clinical setting, there are still uncertainties about the relevance of CSF GAG levels pre- and post-therapy. For example, as the CSF volume increases and brain tissue atrophies in advanced disease, measured CSF GAG levels may artifactually decrease, while clinical manifestations of the disease remain unaffected by treatment.
In the therapeutic development for any disease, dose selection and justification are a fundamental requirement. Of benefit, enzyme replacement therapies often involve no dose or concentration-related safety concerns; immunogenicity to a protein deemed foreign by the patient (due to missing endogenous enzyme) is the main safety concern across enzyme replacement therapies. This affords a unique dosing framework that may not be feasible in other disease settings. In the rare disease setting, in contrast to common diseases, limited dose ranging can be conducted in clinical studies simply due to the small patient population. In particular for lysosomal storage diseases with neurological manifestations, the population with severe disease is primarily pediatric, and enrolling a pediatric patient in a clinical trial should ethically involve a prospect for direct benefit. Therefore, we propose a few principles to guide dose selection through the drug development process.
• Aim to maximize pharmacological response in every individual, rather than mean responses. This is a departure from viewing individual outliers as “outliers”, but rather viewing the individual as less sensitive or least-responder. In this case, theoretically increasing the dose will allow these least-responders to be adequately treated. A responder analysis is routinely applied in common diseases, and while the data available in rare diseases is by definition limited, selecting a dose based on such least-responders is a simple approach to ensure each patient is given the chance of treatment benefit that he or she deserves. Given the single-dose nature of gene therapies, an even more personalized dosing paradigm can be imagined. In principle, doses selected should be those anticipated to be the most efficacious in all individuals, rather than the lowest dose likely to show benefit.
• Aim for normalization of biomarkers (i.e., GAG levels or downstream neurological function markers) as the relationship between substrate reduction and clinical efficacy is unknown. This is fundamentally challenged in pediatric indications by the lack of healthy human pediatric CSF and assays to measure GAG levels. Young children generally only have CSF drawn in the case of suspected infection or injury, which may confound biomarker interpretation. Similarly, the healthy levels of some subspecies of GAGs are so low that they approach the limits of sensitivity of even modern assays. Despite these limitations, normalization provides a clear target for dose selection in the face of complex and not fully understood neurological processes. In some cases, normalization of biomarkers may be approached by administering the maximum feasible dose, based on infusion conditions, drug concentration, etc.
• Leverage animal models of disease to guide dose selection, enabled by the strong evolutionary conservation of lysosomal enzymes. For the family of lysosomal storage diseases, mouse, cat, and/or dog models of disease that are also missing functional enzyme are commonly used for nonclinical pharmacology and toxicology studies. While behavioral or cognitive manifestations of disease in animals may not exactly represent the clinical presentation, biomarkers, including GAGs and other lysosomal markers, often do represent the clinical phenotype. Therefore, once a dose in an animal model has been found to normalize and/or maximize substrate reduction, a clinical dose may be selected. While numerous approaches exist for allometric scaling from animals to humans, scaling based on brain mass is supported for administration routes directly into the CSF [7,8]. In detail, because drug effect is dependent upon enzyme uptake into an amount of brain tissue, scaling should ensure a dose is selected to provide sufficient enzyme to each unit of brain tissue in the patient (and selecting a dose to treat the least well-corrected brain region or cell type). Importantly, dose selection may be confounded in clinical dose-ranging studies by the small number of patients in each dose cohort (as necessitated in a rare disease setting), such that a sub-optimal dose may be selected if results from animal dose-response studies are not considered.
It is heartening to see the number of therapeutics in development for the treatment of the neurological manifestations of lysosomal storage diseases. It is our hope that the framework provided here and exemplified in Grover et al. study [1] facilitates drug development from the preclinical to clinical stages, and that it provides a strong foundation for clinical trial success in these disease states. As noted above, except for Brineura® for the treatment of CLN2, there are no approved treatments for the neurological manifestations of lysosomal storage diseases, and many treatments have failed in development due to lack of demonstration of sufficient efficacy. Therefore, shared learnings to researchers working in such rare, pediatric diseases include the vital importance of the translational stage of drug development to support both treatment safety and efficacy in patients.
References
2. Cohen-Pfeffer JL, Gururangan S, Lester T, Lim DA, Shaywitz AJ, Westphal M, et al. Intracerebroventricular delivery as a safe, long-term route of drug administration. Pediatric Neurology. 2017 Feb 1;67:23-35.
3. Aoyagi-Scharber M, Crippen-Harmon D, Lawrence R, Vincelette J, Yogalingam G, Prill H, et al. Clearance of heparan sulfate and attenuation of CNS pathology by intracerebroventricular BMN 250 in Sanfilippo type B mice. Molecular Therapy-Methods & Clinical Development. 2017 Sep 15;6:43-53.
4. Ullman JC, Nugent AA, Arguello A, Bhalla A, Sandmann T, Lianoglou S, et al. Novel FACS based method demonstrates CNS cell-type distribution and efficacy of a BBB penetrant ERT in a mouse model of MPS II. Molecular Genetics and Metabolism. 2020 Feb 1; 129(2):S155.
5. Vuillemenot BR, Kennedy D, Reed RP, Boyd RB, Butt MT, Musson DG, et al. Recombinant human tripeptidyl peptidase-1 infusion to the monkey CNS: safety, pharmacokinetics, and distribution. Toxicology and Applied Pharmacology. 2014 May 15; 277(1):49-57.
6. Ellinwood NM, Valentine B, Hess AS, Jens JK, Snella EM, Ware WA, et al. Preliminary findings of a twenty-six week or longer intracerebroventricular infusion study of BMN 250 administered once every 2 weeks in a canine model of mucopolysaccharidosis type IIIB. Molecular Genetics and Metabolism. 2017;1(120):S44.
7. Grover A, McCullagh E, Aoyagi-Scharber M, Maricich SM, Wait JC, Melton A, et al. Translational dose-response and frequency scaling for BMN 250 administered via the intracerebroventricular route: Predicting a clinically effective dosing regimen from animal models of disease for the treatment of Sanfilippo syndrome type B. Molecular Genetics and Metabolism. 2018 Feb 1;123(2):S56.
8. Vuillemenot BR, Korte S, Wright TL, Adams EL, Boyd RB, Butt MT. Safety evaluation of CNS administered biologics—study design, data interpretation, and translation to the clinic. Toxicological Sciences. 2016 Jul 1; 152(1):3-9.