Abstract
The two receptors for the anaphylatoxin C5a are critically involved in the recruitment of immune cells and activate these cells at sites of inflammation. The pro-inflammatory function of C5aR1 in these processes is well established, whereas the functional properties of the second C5a receptor, C5aR2, in inflammation remain enigmatic. We recently reported a pro-inflammatory contribution of C5aR2 to the pathogenesis of the prototypical autoimmune skin blistering disease epidermolysis bullosa acquisita (EBA). Deficiency of C5aR2 ameliorated the disease phenotype in an antibody transfer model of EBA and reduced neutrophil migration and activation in vitro. Here, we discuss not only these data, but the crosstalk of C5aR2 with Fcγ receptors, and the effect of C5adesArg stimulation on neutrophils. In addition, we highlight the cellular location of C5aR2, its functional dependence on concomitant C5aR1 expression, and its importance for therapeutic strategies targeting the C5a receptor pathways in neutrophil-mediated diseases.
Keywords
C5aR2, C5a, C5adesArg, Neutrophil, Heterodimerization, Autoimmunity
Introduction
C5aR2 serves as the second receptor for the anaphylatoxin C5a. It was identified about 10 years after identification of the first cognate receptor, C5aR1. Initially, C5aR2 was considered a mere decoy receptor for C5a. According to this view, its function was to scavenge excess C5a from C5aR1 and thereby exert anti-inflammatory effects. However, this initial view of C5aR2 had been oversimplified. Several reports were published pointing toward the functional relevance of this receptor in cell homeostasis as well as in inflammatory conditions driven by immune cells that express both C5a receptors. Importantly, the functions of C5aR2 in such diseases varied substantially from anti- to pro-inflammatory effects and appeared to depend on the cellular and spatial expression of this receptor. Using C5ar2–/– mice, we recently demonstrated a pro-inflammatory contribution of C5aR2 to the pathogenesis of epidermolysis bullosa acquisita (EBA). EBA is a rare autoimmune disease of the skin in which autoantibodies to type VII collagen cause neutrophil-mediated destruction of the dermal-epidermal junction, leading to subepidermal blistering and inflammation. A critical role of C5aR1 in the development and progression of EBA has been reported previously. Mice with a targeted deletion of the C5ar1 gene were almost completely protected from the development of skin lesions and inflammation [1,2]. This protection from disease development was associated with reduced neutrophil function and migration. Because neutrophils are the major effector cells in EBA and express both C5a receptors at high levels, we additionally examined the effects of deleting C5ar2 on C5a-mediated neutrophil function and migration in vitro. We found significantly impaired function and migration of neutrophils from C5ar2–/– mice compared with corresponding cells from wild-type mice. Interestingly, these C5a-mediated functions were entirely dependent on the presence of C5aR1.
The Enigmatic C5aR2
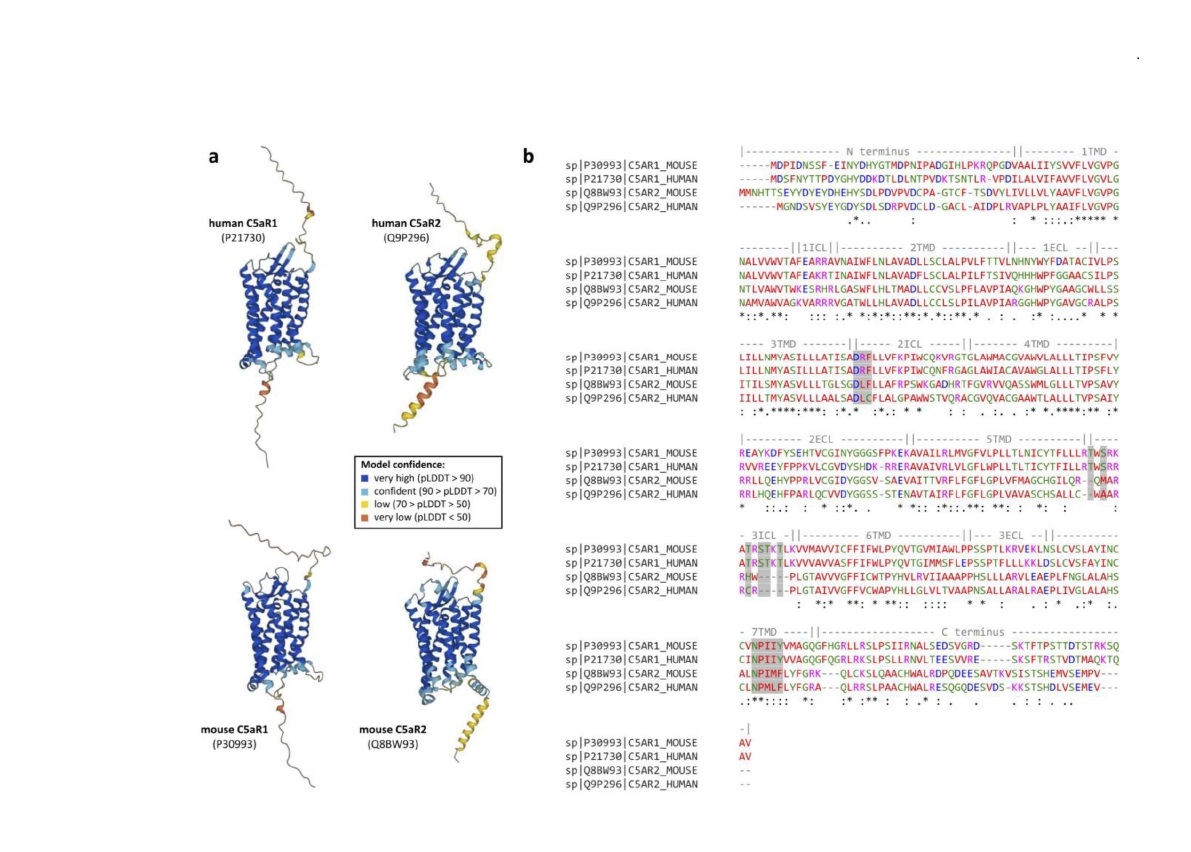
In humans, C5aR2 is abundantly expressed in immune cells such as myeloid cells, natural killer cells, and some T cells [3-8]. In mice, the C5ar2 gene is located on chromosome 7 adjacent to its paralog C5ar1 and encodes for a 344-amino acid seven-transmembrane receptor. Using the recently established floxed tandem dye (td) Tomato-C5ar2 knock-in reporter mouse, C5aR2 has been shown to have an expression pattern comparable to humans in mice, but without a positive signal for C5aR2 in T cells [6,9,10]. For a detailed overview of the expression patterns of C5aR1 and C5aR2 in mice and humans, see the review by Laumonnier et al. [11]. Like C5aR1, C5aR2 contains a potential N-linked glycosylation site at Asn3 and several serine and threonine residues at the C-terminal domain that could serve as phosphorylation sites for GPCR kinases [6,12]. However, despite these possible phosphorylation sites and the general seven-transmembrane structure of C5aR2 (Figure 1a), C5aR2 does not bind to G proteins. This is in part due to (I) a change in the DRY motif, in which the arginine residue important for Gα-protein coupling has been replaced by a leucine residue, (II) missing serine/threonine residues in the third intracellular loop, which are important for G protein recognition in C5aR1, and (III) a change in the NPXXY motif of the seventh transmembrane helix, which functions as an important signal transduction sequence in GPCRs (Figure 1b) [6,7,13-16]. The fact that C5aR2 can bind C5a and its degradation product C5adesArg without triggering G protein-dependent signaling had led to the view of C5aR2 as a decoy receptor acting as a regulator of C5a and C5adesArg availability for C5aR1. However, C5aR2 has been shown to recruit β-arrestins and modulate the recruitment of β-arrestins to C5aR1 [17-19]. Further, several studies have shown that C5aR2 has several functional properties. Interestingly, both pro-inflammatory and anti-inflammatory properties have been reported for C5aR2 – for a detailed overview of the conflicting roles of C5aR2 in pathophysiology, see the review by Li et al. [6]. Anti-inflammatory functions of C5aR2 have been demonstrated in studies of LPS- [20] and IC-mediated lung injury [21], allergic contact dermatitis [22], and recently also in a model of intestinal ischemia-reperfusion injury [23]. On the contrary, C5aR2 exerted pro-inflammatory function in experimental sepsis [24], experimental allergic asthma [25], thioglycolate-induced peritonitis and air-pouch inflammation [26], and surprisingly renal ischemia-reperfusion injury [27,28], and passive EBA [29]. In most cases, C5aR2 acted in concert with C5aR1, suggesting that C5a receptor crosstalk is required for the full development of certain pathological conditions.
Our previously published in vitro results are consistent with this perception of C5aR2 being dependent on the presence of C5aR1, as neutrophil activation, e.g., measured by the increase in intracellular calcium concentration and the up-regulation of CD11b, was completely absent when neutrophils lacked C5aR1. In support we found that pharmacological targeting by the C5aR1 antagonist PMX53 [30-32] or the dual C5aR1/C5aR2 inhibitor A8Δ71-73, abolished C5a-mediated activation of mouse neutrophils (unpublished observation). Such dependence of C5aR2 function on the presence of C5aR1 might be partially explained by the heterodimerization between the two C5a receptors as described previously [4,5,33,34]. It has been reported that heterodimer formation is required for C5a-induced internalization of C5aR1 and downstream PI3K/ERK signaling in a clathrin adaptor protein complex 2 (AP-2)-dependent manner [33].
Of note, to elicit optimal immune responses it also has been shown that complement receptors interact with pattern recognition receptors, particularly TLRs [6,35,36]. Such TLR activation has been shown to enhance C5a-induced pro- inflammatory responses such as the release of IL-6 and IL-8 in the absence of C5a-induced Ca2+ mobilization through negative modulation of C5aR2 [37]. On the other hand, this down-regulation of C5aR2 also negatively affected LPS-induced production of the nuclear protein high mobility group box 1 (HMGB1), an important mediator of inflammation in sepsis. Importantly, the release of HMGB1 is associated with PI3K/Akt activation and depends on C5aR2 but not C5aR1 [6,24,38,39]. In addition, C5aR2 can signal independently of C5aR1 through β-arrestin-scaffolded kinases [6,40], which is why it also has been proposed as an arrestin coupling receptor (ACR) [40].
The Impact of C5aR2 on the C5a-/Fcγ-receptor Crosstalk
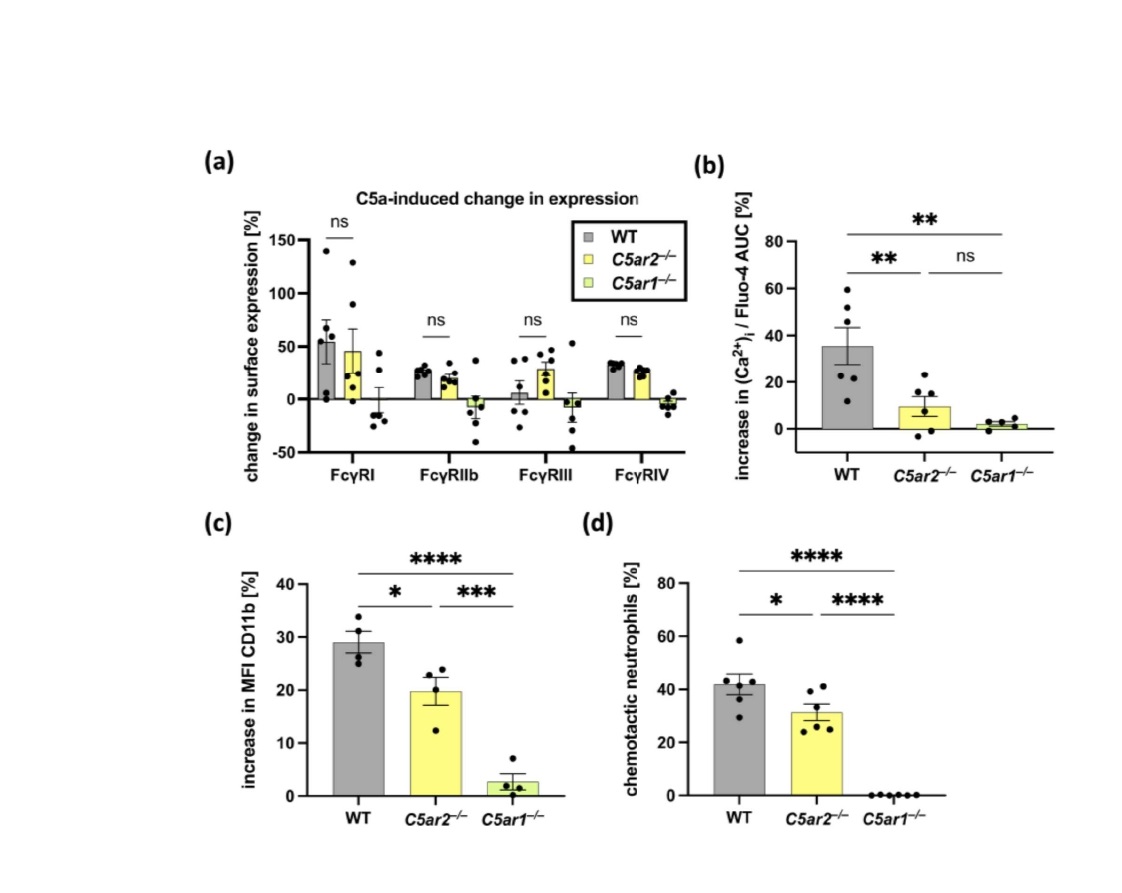
In the past the critical bidirectional interaction between C5a and FcγR activation on cells expressing both receptor families, including neutrophils has been described. Accordingly, C5a generated as a result of immune complex-mediated cellular activation can lead to up-regulation of activating FcγRs and down-regulation of inhibitory FcγRIIb on myeloid cells [1,41-43]. The resulting change in the A/I ratio toward the activating phenotype primes the cells for an inflammatory response through a self-reinforcing feedback loop [29,42,44-46]. With our recently published data, we provide evidence that C5aR2 is involved in regulating the expression of FcγRs on neutrophils and thereby modulates the A/I ratio. In further experiments, we stimulated bone marrow-derived neutrophils with C5a and analyzed the effects on the expression of FcγRs. We found that C5a stimulation led to up-regulation of all FcγRs, including FcγRIIb. Interestingly, although we did not detect a significant difference between FcγR expression in neutrophils from WT and C5ar2–/– mice in response to C5a stimulation, similar to other C5a-mediated neutrophil responses analyzed, we noted that this up-regulation appeared to be dependent on the presence of C5aR1 (Figure 2a). This finding may suggest that the regulation of the A/I ratio by C5aR2 is an indirect effect due to modulation of C5aR1 signaling and does not act directly on FcγRs.
Neutrophils Stimulated with C5adesArg Show Similarly Diminished Responses when C5aR2 is Lacking
An important factor not addressed in our recent publication is the reported higher affinity of C5aR2 for C5adesArg compared with C5aR1. C5adesArg is the desarginated form of C5a, which is the primary degradation product of C5a in response to serum carboxypeptidase N (SCPN) with often reduced biological activity at C5aR1. However, C5adesArg can induce several of the biological activities as C5a when administered at higher concentrations [5,16,47,48]. The rapid degradation of C5a to C5adesArg in serum by SCPN is one mechanism to protect cells from uncontrolled C5aR1 activation, in particular on neutrophils, which are the most abundant leukocyte population in circulation and express high numbers of C5aR1 [5].
To evaluate the impact of C5adesArg on neutrophil activation, we repeated the in vitro neutrophil stimulation experiments presented in Seiler et al., 2022 [29] using C5adesArg as a stimulant. We found that although C5adesArg induced neutrophil chemotaxis and activation, the overall response to C5adesArg was reduced compared to C5a, as expected. However, the effects of reduced neutrophil responsiveness in the absence of C5aR2 that we recently reported after stimulation with C5a were also observed after stimulation with C5adesArg. Moreover, C5adesArg -triggered neutrophil activation and chemotaxis were completely absent when C5aR1 was missing (Figure 2b-d), confirming the important role of C5aR1 in C5a/C5adesArg -driven neutrophil function.
Is C5aR2 an Intracellular Modulator of C5aR1 Signaling in Neutrophils?
One of the several controversies about C5aR2 relates to its cellular expression. Some studies found that only a small fraction (< 20%) of C5aR2 is located on the cell surface while most C5aR2 is predominantly localized within the cell [4,14]. In addition, BRET assays proposed that C5aR1 and C5aR2 form C5a-induced heterodimers, thereby regulating C5a signaling responses [5]. This heterodimerization required high C5a concentrations that are not present in the circulation under physiologic conditions but could be present locally under inflammatory conditions associated with strong complement activation such as in immune complex-driven diseases like EBA.
Together with our previously published data, which demonstrated the dependence of C5aR2 function on the presence of C5aR1 in neutrophils, we propose that C5a is made available to the large pool of intracellular C5aR2 after binding of C5a to C5aR1 and subsequent internalization, which may be dependent on surface expressed C5aR2 [33]. In turn, intracellular C5aR2 may influence the function of C5aR1, either by triggering a signaling cascade itself or by transporting C5a or C5aR1 back to the cell surface to initiate repeated C5a/C5aR1-signaling [6]. This view is consistent with the observation that C5a, but not C5adesArg, induces the formation of C5aR1/2 heterodimers [5]. Therefore, together with the lower affinity of C5adesArg for C5aR1, this could also contribute to the lower overall neutrophil response to C5adesArg compared to C5a.
Of note, based on its intracellular localization, C5aR2 may also contribute to immunometabolism, i.e., the impact of immune functions on cell metabolism [16,34,49,50].
C5a Receptor Targeting in Neutrophil-driven Diseases
The recent FDA approval of several compounds that target the complement system either at the level of the classical pathway (CP) [51,52], the alternative pathway (AP) C3 [53,54], or the terminal pathway C5aR1 [55] and the successful treatment of paroxysmal nocturnal hemoglubinuria (PNH) patients with the terminal pathway inhibiting anti-C5 mAb eculizumab and derivatives [56-58] paved the way for an extensive pipeline of new therapeutic inhibitors targeting the complement system at several levels [59-61].
While specific blockade of one activation pathway might be an appropriate strategy to inhibit adverse complement activation, in some complement-related diseases such as cold agglutinin disease (CP), IgA nephropathy and certain thrombotic microangiopathies (lectin pathway, LP) as well as primary membranous nephropathy (AP), C5a and the membrane-attack complex formation, as effectors of the terminal pathway, critically contribute to the clinical manifestation of several complement-mediated diseases such as PNH, atypical hemolytic uremic syndrome (aHUS) or antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis [62]. Clearly, blocking C5a or C5a receptors might serve as an important immune target in neutrophil-mediated diseases, in particular in response to immune complex-driven diseases in the skin such as bullous pemphigoid diseases including EBA.
Due to the overwhelming evidence of C5aR1 as the main effector molecule of C5a/C5adesArg-induced activation of immune cells and the still controversial and enigmatic role of C5aR2, C5aR1 has been considered the main C5aR target. Recently, avacopan (CCX168, Tavneos, ChemoCentryx), a small molecule that effectively blocks C5aR1 but not C5aR2, has been approved for the treatment of ANCA-associated vasculitis [55]. While the specific targeting of C5aR1 in this case was justified with findings from mouse studies of anti-myeloperoxidase (MPO)-induced necrotizing and crescentic glomerulonephritis that suggested a pro-inflammatory contribution of C5aR1 but a suppressive or anti-inflammatory mode of action for C5aR2 [63], our recent study clearly demonstrated that C5aR2 can also contribute to neutrophil-driven diseases following complement activation. Accordingly, therapeutic targeting of C5aR2 in addition to C5aR1 might be a strategy to consider in defined clinical settings such as autoimmune blistering of the skin or in severe sepsis [64,65].
Given the widespread expression of C5aR2 in various immune (e.g., granulocytes) and non-immune (e.g., cardiomyocytes) cells, it is reasonable to speculate that C5aR2 may have different functions depending on the tissue, cell, and/or subcellular location, which should be considered for therapeutic modulation of this receptor. Moreover, therapeutic targeting of C5aR2, in contrast to C5aR1, may pose particular challenges to be addressed due to its predominantly intracellular location. Although there are compounds that have been reported to have both binding and functional activity at C5aR2, to the best of our knowledge only the linear peptides P32 and P59 [66] are specific (agonists) for C5aR2, and a specific receptor antagonist has yet to be discovered. Clearly, such discoveries then need to be tested experimentally in appropriate animal models to get a better understanding of C5aR2 function in the specific disease setting.
Of note, although C5a and C5aRs are well conserved across species, there are reports of defined differences between mouse and human C5a and C5aRs (e.g., [67]). According to this, both human C5a (hC5a) and murine C5a (mC5a) act as C5aRs agonists regardless of the receptor species, with mC5a exhibiting higher potency. The degradation product hC5adesArg is a partial agonist for hC5aRs and, to a lesser extent, for mC5aRs. In contrast, mC5adesArg is a partial agonist of hC5aRs but has full agonist potency for mC5aRs, albeit reduced compared to mC5a. Therefore, it will be crucial to verify the functions of C5aR2 found in animal models – such as the net pro-inflammatory contribution of C5aR2 in antibody-transfer experimental EBA, which we have previously reported – in humans to draw accurate conclusions for its clinical use.
Conclusion
Our recent publication highlighted the critical function of C5aR2 in EBA disease development and showed a clear pro-inflammatory contribution. In neutrophils, which are critical drivers of inflammation in EBA, these effects were associated with and depending on C5aR1. Our study also showed that C5aR2 can affect the expression of other receptors, such as FcγRs, whose activation is involved in innate effector functions, i.e., ROS release. At this point, the underlying molecular signals downstream of C5aR2 activation are still unknown as is the potential of C5aR2 to dimerize with other seven-transmembrane receptors. Clearly, further research is required to uncover the signaling pathways of C5aR2 in different immune cells and in different inflammatory conditions and its crosstalk with other receptors at the molecular level. Such knowledge will fuel our understanding of when and where C5aR2 targeting might prove useful. This could be of particular relevance in neutrophils, which are not only involved in innate host defense but play an important role as drivers of autoimmune diseases such as ANCA-associated vasculitis or skin blistering diseases.
Material & Methods
Mice
C57BL/6J (WT) as well as C5ar1–/– and C5ar2–/– mice on the C57BL/6J genetic background were bred and housed in a 12-hour light-dark cycle at the University of Lübeck animal facility (Lübeck, Germany).
Bone marrow cell preparation
Isolation of bone marrow cells from femurs and tibiae is described in Seiler et al., 2022 [29].
C5a-induced changes in surface expression of FcγRs on neutrophils
To investigate the influence of C5a stimulation on the surface expression of FcγRs on neutrophils, bone marrow cells from C57BL/6J (WT), C5ar1–/– and C5ar2–/– (both on C57BL/6J background) mice were isolated as described [29]. Isolated bone marrow cells were incubated in complete RPMI-1640 medium containing 12.5 nM C5a or no C5a as a control for 30 minutes at 37°C, 5% carbon dioxide. Cells were washed and stained for Ly6G, CD11b, FcγRI, FcγRIIb, FcγRIII, and FcγRIV. Surface expression of FcγRs on C5a-stimulated and nonstimulated Ly6G+/CD11b+ cells were determined using a BD LSRII flow cytometer. Changes in FcγRs expression were quantified by calculating the relative increase in mean fluorescence intensity of the FcγRI, FcγRIIb, FcγRIII, or FcγRIV signal in response to C5a stimulation.
Assessment of (Ca2+)i changes in bone marrow neutrophils
Flow cytometry-based analysis of intracellular calcium flux is described in Seiler et al., 2022 [29].
CD11b up-regulation
The assessment of C5a-induced CD11b up-regulation is described in Seiler et al., 2022 [29].
Acknowledgements
We acknowledge financial support by Land SchleswigHolstein within the funding program Open Access Publikationsfonds.
References
2. Mihai S, Hirose M, Wang Y, Thurman JM, Holers VM, Morgan BP, et al. Specific inhibition of complement activation significantly ameliorates autoimmune blistering disease in mice. Frontiers in Immunology. 2018 Mar 16;9:535.
3. Arbore G, West EE, Spolski R, Robertson AA, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4+ T cells. Science. 2016 Jun 17;352(6292):aad1210.
4. Bamberg CE, Mackay CR, Lee H, Zahra D, Jackson J, Lim YS, et al. The C5a receptor (C5aR) C5L2 is a modulator of C5aR-mediated signal transduction. Journal of Biological Chemistry. 2010 Mar 5;285(10):7633-44.
5. Croker DE, Halai R, Fairlie DP, Cooper MA. C5a, but not C5a‐des Arg, induces upregulation of heteromer formation between complement C5a receptors C5aR and C5L2. Immunology and Cell Biology. 2013 Nov;91(10):625-33.
6. Li XX, Lee JD, Kemper C, Woodruff TM. The complement receptor C5aR2: a powerful modulator of innate and adaptive immunity. The Journal of Immunology. 2019 Jun 15;202(12):3339-48.
7. Okinaga S, Slattery D, Humbles A, Zsengeller Z, Morteau O, Kinrade MB, et al. C5L2, a nonsignaling C5A binding protein. Biochemistry. 2003 Aug 12;42(31):9406-15.
8. Pundir P, MacDonald CA, Kulka M. The novel receptor C5aR2 is required for C5a-mediated human mast cell adhesion, migration, and proinflammatory mediator production. The Journal of Immunology. 2015 Sep 15;195(6):2774-87.
9. Karsten CM, Wiese AV, Mey F, Figge J, Woodruff TM, Reuter T, et al. Monitoring C5aR2 expression using a floxed tdTomato-C5aR2 knock-in mouse. The Journal of Immunology. 2017 Nov 1;199(9):3234-48.
10. Verghese DA, Demir M, Chun N, Fribourg M, Cravedi P, Llaudo I, et al. T cell expression of C5a receptor 2 augments murine regulatory T cell (TREG) generation and TREG-dependent cardiac allograft survival. The Journal of Immunology. 2018 Mar 15;200(6):2186-98.
11. Laumonnier Y, Karsten CM, Köhl J. Novel insights into the expression pattern of anaphylatoxin receptors in mice and men. Molecular Immunology. 2017 Sep 1;89:44-58.
12. Ohno M, Hirata T, Enomoto M, Araki T, Ishimaru H, Takahashi TA. A putative chemoattractant receptor, C5L2, is expressed in granulocyte and immature dendritic cells, but not in mature dendritic cells. Molecular Immunology. 2000 Jun 1;37(8):407-12.
13. He R, Browning DD, Richard DY. Differential roles of the NPXXY motif in formyl peptide receptor signaling. The Journal of Immunology. 2001 Mar 15;166(6):4099-105.
14. Scola AM, Johswich KO, Morgan BP, Klos A, Monk PN. The human complement fragment receptor, C5L2, is a recycling decoy receptor. Molecular Immunology. 2009 Mar 1;46(6):1149-62.
15. Scola AM, Higginbottom A, Partridge LJ, Reid RC, Woodruff T, Taylor SM, et al. The role of the N-terminal domain of the complement fragment receptor C5L2 in ligand binding. Journal of Biological Chemistry. 2007 Feb 9;282(6):3664-71.
16. Zhang T, Garstka MA, Li K. The controversial C5a receptor C5aR2: its role in health and disease. Journal of Immunology Research. 2017 Oct;2017.
17. Braun L, Christophe T, Boulay F. Phosphorylation of key serine residues is required for internalization of the complement 5a (C5a) anaphylatoxin receptor via a β-arrestin, dynamin, and clathrin-dependent pathway. Journal of Biological Chemistry. 2003 Feb 7;278(6):4277-85.
18. Croker DE, Halai R, Kaeslin G, Wende E, Fehlhaber B, Klos A, et al. C5a2 can modulate ERK1/2 signaling in macrophages via heteromer formation with C5a1 and β‐arrestin recruitment. Immunology and Cell Biology. 2014 Aug;92(7):631-9.
19. Kalant D, MacLaren R, Cui W, Samanta R, Monk PN, Laporte SA, et al. C5L2 is a functional receptor for acylation-stimulating protein. Journal of Biological Chemistry. 2005 Jun 24;280(25):23936-44.
20. Wang R, Lu B, Gerard C, Gerard NP. C5L2, the second C5a anaphylatoxin receptor, suppresses LPS-induced acute lung injury. American Journal of Respiratory Cell and Molecular Biology. 2016 Nov;55(5):657-66.
21. Gerard NP, Lu B, Liu P, Craig S, Fujiwara Y, Okinaga S, et al. An anti-inflammatory function for the complement anaphylatoxin C5a-binding protein, C5L2. Journal of Biological Chemistry. 2005 Dec 2;280(48):39677-80.
22. Wang R, Lu B, Gerard C, Gerard NP. Disruption of the complement anaphylatoxin receptor C5L2 exacerbates inflammation in allergic contact dermatitis. The Journal of Immunology. 2013 Oct 15;191(8):4001-9.
23. Wu MC, Lee JD, Ruitenberg MJ, Woodruff TM. Absence of the C5a receptor C5aR2 worsens ischemic tissue injury by increasing C5aR1-mediated neutrophil infiltration. The Journal of Immunology. 2020 Nov 15;205(10):2834-9.
24. Rittirsch D, Flierl MA, Nadeau BA, Day DE, Huber-Lang M, Mackay CR, et al. Functional roles for C5a receptors in sepsis. Nature Medicine. 2008 May;14(5):551-7.
25. Zhang X, Schmudde I, Laumonnier Y, Pandey MK, Clark JR, König P, Gerard NP, Gerard C, Wills-Karp M, Köhl J. A critical role for C5L2 in the pathogenesis of experimental allergic asthma. The Journal of Immunology. 2010 Dec 1;185(11):6741-52.
26. Chen NJ, Mirtsos C, Suh D, Lu YC, Lin WJ, McKerlie C, et al. C5L2 is critical for the biological activities of the anaphylatoxins C5a and C3a. Nature. 2007 Mar;446(7132):203-7.
27. Poppelaars F, van Werkhoven MB, Kotimaa J, Veldhuis ZJ, Ausema A, Broeren SG, et al. Critical role for complement receptor C5aR2 in the pathogenesis of renal ischemia‐reperfusion injury. The FASEB Journal. 2017 Jul;31(7):3193-204.
28. Thorenz A, Derlin K, Schröder C, Dressler L, Vijayan V, Pradhan P, et al. Enhanced activation of interleukin-10, heme oxygenase-1, and AKT in C5aR2-deficient mice is associated with protection from ischemia reperfusion injury–induced inflammation and fibrosis. Kidney International. 2018 Oct 1;94(4):741-55.
29. Seiler DL, Kleingarn M, Kähler KH, Gruner C, Schanzenbacher J, Ehlers-Jeske E, et al. C5aR2 Deficiency Ameliorates Inflammation in Murine Epidermolysis Bullosa Acquisita by Regulating Fcγ Receptor Expression on Neutrophils. Journal of Investigative Dermatology. 2022 Oct 1;142(10):2715-23.
30. Finch AM, Wong AK, Paczkowski NJ, Wadi SK, Craik DJ, Fairlie DP, et al. Low-molecular-weight peptidic and cyclic antagonists of the receptor for the complement factor C5a. Journal of Medicinal Chemistry. 1999 Jun 3;42(11):1965-74.
31. Morgan M, Bulmer AC, Woodruff TM, Proctor LM, Williams HM, Stocks SZ, et al. Pharmacokinetics of a C5a receptor antagonist in the rat after different sites of enteral administration. European Journal of Pharmaceutical Sciences. 2008 Apr 23;33(4-5):390-8.
32. Strachan AJ, Woodruff TM, Haaima G, Fairlie DP, Taylor SM. A new small molecule C5a receptor antagonist inhibits the reverse-passive Arthus reaction and endotoxic shock in rats. The Journal of Immunology. 2000 Jun 15;164(12):6560-5.
33. Hsu WC, Yang FC, Lin CH, Hsieh SL, Chen NJ. C5L2 is required for C5a-triggered receptor internalization and ERK signaling. Cellular Signalling. 2014 Jul 1;26(7):1409-19.
34. Poursharifi P, Lapointe M, Pétrin D, Devost D, Gauvreau D, Hébert TE, et al. C5L2 and C5aR interaction in adipocytes and macrophages: insights into adipoimmunology. Cellular Signalling. 2013 Apr 1;25(4):910-8.
35. Hajishengallis G, Lambris JD. More than complementing Tolls: complement–Toll‐like receptor synergy and crosstalk in innate immunity and inflammation. Immunological Reviews. 2016 Nov;274(1):233-44.
36. Hawlisch H, Belkaid Y, Baelder R, Hildeman D, Gerard C, Köhl J. C5a negatively regulates toll-like receptor 4-induced immune responses. Immunity. 2005 Apr 1;22(4):415-26.
37. Raby AC, Holst B, Davies J, Colmont C, Laumonnier Y, Coles B, et al. TLR activation enhances C5a‐induced pro‐inflammatory responses by negatively modulating the second C5a receptor, C5L2. European Journal of Immunology. 2011 Sep;41(9):2741-52.
38. Gardella S, Andrei C, Ferrera D, Lotti LV, Torrisi MR, Bianchi ME, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Reports. 2002 Oct 1;3(10):995-1001.
39. Wang H, Zhu S, Zhou R, Li W, Sama AE. Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Reviews in Molecular Medicine. 2008 Nov;10.
40. Pandey S, Kumari P, Baidya M, Kise R, Cao Y, Dwivedi-Agnihotri H, Banerjee R, Li XX, Cui CS, Lee JD, Kawakami K. Intrinsic bias at non-canonical, β-arrestin-coupled seven transmembrane receptors. Molecular Cell. 2021 Nov 18;81(22):4605-21.
41. Godau J, Heller T, Hawlisch H, Trappe M, Howells E, Best J, et al. C5a initiates the inflammatory cascade in immune complex peritonitis. The Journal of Immunology. 2004 Sep 1;173(5):3437-45.
42. Kumar V, Ali SR, Konrad S, Zwirner J, Verbeek JS, Schmidt RE, et al. Cell-derived anaphylatoxins as key mediators of antibody-dependent type II autoimmunity in mice. The Journal of clinical investigation. 2006 Feb 1;116(2):512-20.
43. Shushakova N, Skokowa J, Schulman J, Baumann U, Zwirner J, Schmidt RE, et al. C5a anaphylatoxin is a major regulator of activating versus inhibitory FcγRs in immune complex–induced lung disease. The Journal of Clinical Investigation. 2002 Dec 15;110(12):1823-30.
44. Atkinson JP. C5a and Fcγ receptors: a mutual admiration society. The Journal of Clinical Investigation. 2006 Feb 1;116(2):304-6.
45. Karsten CM, Köhl J. The immunoglobulin, IgG Fc receptor and complement triangle in autoimmune diseases. Immunobiology. 2012 Nov 1;217(11):1067-79.
46. Schmidt RE, Gessner JE. Fc receptors and their interaction with complement in autoimmunity. Immunology Letters. 2005 Aug 15;100(1):56-67.
47. Monk PN, Scola AM, Madala P, Fairlie DP. Function, structure and therapeutic potential of complement C5a receptors. British Journal of Pharmacology. 2007 Oct;152(4):429-48.
48. Reis ES, Chen H, Sfyroera G, Monk PN, Köhl J, Ricklin D, Lambris JD. C5a receptor-dependent cell activation by physiological concentrations of desarginated C5a: insights from a novel label-free cellular assay. The Journal of Immunology. 2012 Nov 15;189(10):4797-805.
49. Arbore G, Kemper C. A novel “complement–metabolism–inflammasome axis” as a key regulator of immune cell effector function. European Journal of Immunology. 2016 Jul;46(7):1563-73.
50. Rezvani R, Smith J, Lapointe M, Marceau P, Tchernof A, Cianflone K. Complement receptors C5aR and C5L2 are associated with metabolic profile, sex hormones, and liver enzymes in obese women pre-and postbariatric surgery. Journal of Obesity. 2014 Jan 1;2014.
51. Röth A, Barcellini W, D'Sa S, Miyakawa Y, Broome CM, Michel M, et al. Sutimlimab in Cold Agglutinin Disease. The New England Journal of Medicine. 2021 Apr 8;384(14):1323-1334.
52. Röth A, Berentsen S, Barcellini W, D'Sa S, Jilma B, Michel M, et al. Sutimlimab in patients with cold agglutinin disease: results of the randomized placebo-controlled phase 3 CADENZA trial. Blood. 2022 Sep 1;140(9):980-991.
53. Hillmen P, Szer J, Weitz I, Röth A, Höchsmann B, Panse J, et al. Pegcetacoplan versus Eculizumab in Paroxysmal Nocturnal Hemoglobinuria. The New England Journal of Medicine. 2021 Mar 18;384(11):1028-1037.
54. Wong RSM, Pullon HWH, Amine I, Bogdanovic A, Deschatelets P, Francois CG, et al. Inhibition of C3 with pegcetacoplan results in normalization of hemolysis markers in paroxysmal nocturnal hemoglobinuria. Annals of Hematology. 2022 Sep;101(9):1971-1986.
55. Jayne DR, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis. New England Journal of Medicine. 2021 Feb 18;384(7):599-609.
56. Hillmen P, Young NS, Schubert J, Brodsky RA, Socié G, Muus P, et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. The New England Journal of Medicine. 2006 Sep 21;355(12):1233-43.
57. Schubert ML, Peura DA. Control of gastric acid secretion in health and disease. Gastroenterology. 2008 Jun;134(7):1842-60.
58. Kulasekararaj AG, Brodsky RA, Hill A. Monitoring of patients with paroxysmal nocturnal hemoglobinuria on a complement inhibitor. American Journal of Hematology. 2021 Jul 1;96(7):E232-E235.
59. Mastellos DC, Ricklin D, Lambris JD. Clinical promise of next-generation complement therapeutics. Nature Reviews Drug Discovery. 2019 Sep;18(9):707-29.
60. Ricklin D, Mastellos DC, Reis ES, Lambris JD. The renaissance of complement therapeutics. Nature Reviews Nephrology. 2018 Jan;14(1):26-47.
61. Ricklin D, Mastellos DC, Lambris JD. Therapeutic targeting of the complement system. Nature Reviews. Drug discovery. 2019 Dec 9.
62. Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nature Reviews Nephrology. 2016 Jul;12(7):383-401.
63. Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a receptor (CD88) blockade protects against MPO-ANCA GN. Journal of the American Society of Nephrology. 2014 Feb 1;25(2):225-31.
64. Ward PA. The harmful role of c5a on innate immunity in sepsis. Journal of Innate Immunity. 2010;2(5):439-45.
65. Grailer JJ, Fattahi F, Dick RS, Zetoune FS, Ward PA. Cutting edge: critical role for C5aRs in the development of septic lymphopenia in mice. The Journal of Immunology. 2015 Feb 1;194(3):868-72.
66. Croker DE, Monk PN, Halai R, Kaeslin G, Schofield Z, Wu MC, et al. Discovery of functionally selective C5aR2 ligands: novel modulators of C5a signalling. Immunology and Cell Biology. 2016 Sep;94(8):787-95.
67. Schatz-Jakobsen JA, Yatime L, Larsen C, Petersen SV, Klos A, Andersen GR. Structural and functional characterization of human and murine C5a anaphylatoxins. Acta Crystallographica Section D: Biological Crystallography. 2014 Jun 1;70(6):1704-17.
68. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021 Aug;596(7873):583-589.
69. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold Protein Structure Database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Research. 2022 Jan 7;50(D1):D439-D444.
70. Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecular Systems Biology. 2011 Oct 11;7:539.