Abstract
Tuberous sclerosis complex (TSC) is a neurocutaneous syndrome characterized by systemic hamartomas, including skin and neural symptoms. Many patients exhibit epilepsy, intellectual disability, autism, and other behavioral and neuropsychiatric symptoms, referred to as TSCassociated neuropsychiatric disorders (TAND). TSC involves the overactivation of the mechanistic target of rapamycin (mTOR), specifically mTOR complex 1 (mTORC1). Therefore, mTORC1 inhibitors effectively treat brain, kidney, lung, and skin hamartomas in TSC. However, the effect of mTORC1 inhibitors on neurological symptoms in patients with TSC remains unclear, despite reports of mTORC1 inhibitors improving epilepsy, behavioral disorders, and learning disabilities in animal models.
In our study, we revealed that MITF-specific TSC2 conditional knockout mice (TSC2 cKO) exhibit epilepsy and TAND phenotypes. Furthermore, mTORC1 inhibitor sirolimus treatment ameliorated epilepsy and some TAND symptoms in TSC2 cKO mice. Neurodegeneration was observed in the brain regions responsible for these symptoms. Neuroinflammation caused by the M1 phenotype of microglia is one of the factors contributing to neurodegeneration. In our study, microglia shifted to the anti-inflammatory M2 phenotype when TSC2 cKO mice were treated with sirolimus. Hyperactivity of mTORC1 inhibits autophagy, and increased autophagy activity polarizes microglia to the M2 phenotype. One of the possible mechanisms by which sirolimus can change microglial polarity is by inhibiting mTORC1 activity, which increases autophagy activity. In addition, sirolimus, which shifts microglial polarity to anti-inflammatory, may effectively treat TSC and other neurodegenerative diseases.
Keywords
mTORC1 inhibitor, Sirolimus, Tuberous sclerosis complex, Microglia polarity, TSC-associated neuropsychiatric disorders, Epilepsy, Neuroinflammation
Introduction
Tuberous sclerosis complex (TSC) and mechanistic target of rapamycin complex 1 (mTORC1) inhibitors
TSC is a neurocutaneous syndrome characterized by hamartomas in the brain, skin, and many other organs [1]. In addition to physical symptoms, epilepsy affects 80% of patients with TSC, and seizures are usually severe and difficult to treat. In addition, many patients with TSC have severe neurodevelopmental disorders, such as intellectual disability, autism, anxiety, attention-deficit hyperactivity disorder (ADHD), and aggressive or disruptive behaviors, which are referred to as TSC-associated neuropsychiatric disorder (TAND) [2-4].
The TuberOus SClerosis registry to increase disease Awareness (TOSCA) reported that the common behavioral problems among patients with TAND include hyperactivity, impulsivity, sleep disturbances, anxiety, mood swings, aggression, depressed mood, self-injurious behavior, and obsessions. Additionally, these mental disorders included 21.1% of autistic spectrum disorder (ASD), 19.1% of ADHD, 9.7% of anxiety disorders, and 6.1% of depressive disorders [5]. In addition, several types of memory deficits have been observed in patients with TSC [6]. These neurological and neuropsychiatric manifestations of TSC are treatment burdens for patients and their families. These manifestations of TSC involve the mechanistic target of rapamycin complex (mTOR) pathway. mTOR functions in two distinct protein complexes called mTORC1 and mTORC2. TSC is triggered by mutations in the TSC1 or TSC2 genes, which encode hamartin or tuberin, respectively; these proteins work together to suppress mTORC1 function. The overactivation of mTORC1 in TSC by mutations in TSC1 or TSC2 promotes cell growth and tumor formation because mTORC1 regulates physiological functions, including cell growth and proliferation, metabolism, and protein synthesis [7]. Therefore, mTORC1 inhibitors, such as sirolimus and everolimus, are effective treatments for brain, kidney, lung, and skin hamartomas in TSC [8,9].
Effects of mTORC1 inhibitors on epilepsy and TAND phenotypes
Animal experiments: In animal models, such as knockout and conditional knockout mice targeting TSC1 or TSC2, epilepsy, neurocognitive defects, learning disability, and autism-like behavioral abnormalities were observed. Furthermore, mTORC1 inhibitors such as sirolimus and everolimus have been reported to improve these symptoms [10-12].
Clinical trials: Compared with the efficacy of mTORC1 inhibitors for brain, kidney, lung, and skin hamartomas in TSC, their efficacy for TSC neuropsychiatric symptoms is minimal. Adjunctive treatment with everolimus is effective for refractory epilepsy in patients with TSC [13]. However, recent trials with everolimus discovered no significant effect on TAND, including cognitive function, ASD, and neuropsychiatric deficits (memory and attention) [14,15]. Moreover, the efficacy of mTORC1 inhibitors in TAND has not been fully evaluated.
Epilepsy, TAND, and Neurodegeneration in TSC Model Mice
Our group generated TSC2flox/flox, Mitf-Cre conditional knockout (TSC2 cKO) mice and performed a detailed behavioral analysis of the cKO mice [16]. These TSC2 cKO mice have been previously reported in our studies to significantly reduce skin pigmentation and exhibit spontaneous epilepsy [17,18]. Furthermore, this detailed behavioral analysis suggested that TSC2 cKO mice may be a useful model for TAND, as they exhibit epilepsy and multiple neuropsychiatric phenotypes: ADHD-like behavior, anxiety-like symptoms, ASD-like behavior, and significant impairment of spatial working memory [16]. Cre expression analysis also revealed that the olfactory bulb, piriform cortex, and hippocampus were the sites responsible for epilepsy and TAND in TSC2 cKO mice, and neurodegeneration occurred in these regions [16]. Intraperitoneal administration of sirolimus to TSC2 cKO mice alleviated epilepsy, DHD-like behavior, anxiety-like symptoms, and spatial working memory deficits, indicating that sirolimus is effective in treating epilepsy and neuropsychiatric symptoms in Tsc2 cKO mice. Furthermore, sirolimus treatment reduced neurodegeneration in the olfactory bulb, piriform cortex, and hippocampus.
In Alzheimer's disease (AD) and Parkinson's disease (PD), typical neurodegenerative diseases, persistent neuroinflammation by microglia is involved in the onset and worsening of these conditions. Furthermore, inflammation is involved in neurological diseases such as epilepsy and Guillain-Barré syndrome [20,21]. Therefore, suppressing neuroinflammation might ameliorate symptoms of neurodegenerative diseases [19].
Introduction of Neuroinflammation
Neurodegeneration is primarily observed in neurodegenerative diseases, such as AD and PD, that adversely affect mental and physical functions. Neuroinflammation is involved in central nervous system (CNS) degeneration. Neuroinflammation is a defense mechanism that maintains tissue homeostasis, but it can cause tissue damage and disease if it is severe or prolonged [22]. Activation of receptors involved in innate immunity also induces inflammation in neuroinflammation.
Induction of inflammation
Pattern recognition receptors (PRRs) that bind to pathogen-associated molecular patterns initiate inflammatory responses to infectious pathogens. PRRs include toll-like receptors, receptors for advanced glycation end products, and C-type lectin receptors. The binding of damage-associated molecular patterns, such as ribonucleic acid released by damaged cells, mitochondrial deoxyribonucleic acid, heat shock proteins, alpha-synuclein, and amyloid-β (Aβ) peptides derived from Aβ protein, to PRRs causes inflammation in the brain, inducing inflammation signals [23]. PRRs are highly expressed in macrophages and microglia. In addition to PRRs, purinergic receptors are expressed in microglia and play an important role in regulating microglial activity in response to adenosine triphosphate (ATP) released from damaged cells.
Inflammation by Microglia
Microglia are nerve-specific immune cells, and their main functions as resident macrophages in the brain are pathogen defense and phagocytosis by constantly investigating the surrounding microenvironment, tissue repair, and neuronal support to maintain CNS homeostasis [24]. Under physiological conditions, microglia exhibit an inactivated phenotype. However, when a change in brain homeostasis is detected, specifically pathogen invasion or tissue damage, microglia switch to an activated phenotype, extending processes toward the injury site and initiating repair by eliminating (clearance) pathogens, dead cells and debris, and abnormal cells [25].
Cell damage due to persistent inflammation
Some inflammatory stimuli have beneficial effects, such as the phagocytosis of cellular debris and apoptotic cells. In most cases, this response resolved as an infection is eradicated, or tissue damage is repaired. However, the inflammatory stimulus may persist when inflammation persists, or the normal mechanisms toward inflammatory convergence may fail, resulting in an uncontrolled inflammatory response that may exacerbate the underlying disease [26]. For example, in the early stages of AD, microglial activation clears Aβ. However, abnormal microglial activation during AD progression produces inflammatory cytokines and triggers inducible nitric oxide synthase expression, which can kill neurons [27]. Thus, the blockade of these responses may ameliorate symptoms of neurodegenerative diseases.
Activated microglial phenotype and its expression markers
Activated microglia are polarized into two distinct phenotypes: M1 and M2. M1-phenotype microglia exhibit inflammation-inducing properties, whereas the M2 phenotype repairs tissue by clearing cellular debris and releasing numerous protective factors, such as anti-inflammatory cytokines and nerve growth factors [28]. Macrophages and microglia can recognize each functional state using specific markers and morphology. M1 phenotypic markers include CD16, CD32, CD86, and inducible nitric oxide synthase. The M2 phenotype has subtypes: M2a, M2b, and M2c. M2 phenotypic markers include arginase-1, Ym1, Fizz, IL-10, MHC II, CD163, and CD206 TGF-β [28]. However, microglia are not strictly polarized into M1 and M2-only phenotypes but are influenced by the environment and shift into a continuous intermediate phenotype between M1 and M2 to maintain tissue homeostasis [29].
Expression of microglial markers in TSC2 cKO mice
In TSC2 cKO olfactory bulb treated with sirolimus, microglial morphology was predominantly larger, and microglial process branching increased [16] (Figures 1A and 1B). Sirolimus treatment resulted in predominantly higher expressions of IL-10, Arg1, and CD206, which are expressed in the M2 phenotype (Figure 1C). In contrast, TSC2 cKO mice without sirolimus treatment had lower IL-10, Arg1, and CD206 expressions, but not predominantly higher expressions of IL-6 and IL-1β (markers of M1 phenotype) and were polarized toward M1 state rather than a complete M1 state, suggesting that sirolimus treatment shifted microglia toward the M2 phenotype.
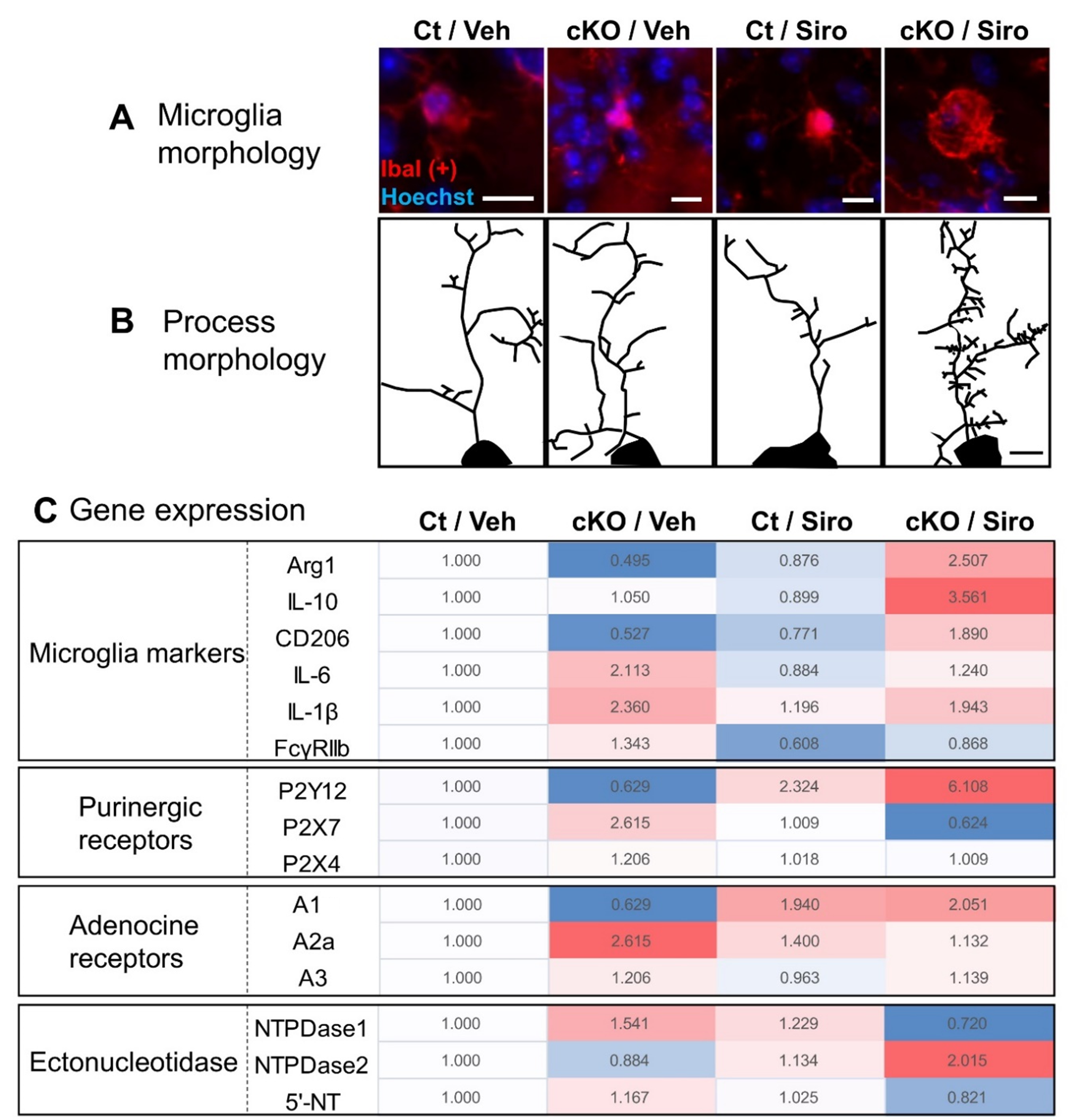
Figure 1. Changes in microglial morphology and gene expression after sirolimus treatment in TSC2 cKO mice. A. Microglial morphology in each treatment group. B. Morphology of microglial processes in each treatment group. C. Comparison of the expression of microglial markers (M2: Arg,1, IL-10, and CD206; M1: IL-6, IL-β, and FcγRIIb), purinergic receptors, and adenosine receptors; and comparison of adenosine receptors and ectonucleotidase expression. The value of vehicle-treated control mice (Ct/Veh) was set at 1.000, and the average of each treatment was color-coded. A darker red indicates a higher expression, and a darker blue indicates a lower expression. Scale bars indicate 10 μm in B and 5 μm in B. A and B were modified from those used in [16].
Purinergic Receptors
Purinergic receptors are expressed in microglia and regulate microglial activity in response to ATP released from damaged cells. Purinergic receptors are classified as P1 and P2 receptors [30]. The P1 receptor is an adenosine-sensitive receptor with four subtypes: A1, A2a, A2b, and A3. P2 receptors, on the other hand, include P2X (P2X1-P2X7) and P2Y (P2Y1, 2, 4, 6, 11, 12, 13, 14) receptors that are activated by the nucleosides adenosine diphosphate (ADP) and ATP and uridine diphosphate and triphosphate, respectively. Purinergic metabolites in the brain constitute important signals that promote microglial activation and chemotaxis [31].
Subsequently, we described P2Y12 and A2a receptors that are markedly altered in the microglia of TSC2 cKO mice after sirolimus treatment.
P2Y12 receptors are highly expressed in microglia and are primarily activated by ADP [32]. P2Y12 receptors are essential for microglial chemotaxis and participate in microglial response [33], neuronal protection [34], and maintenance of the blood-brain barrier [35]. The P2Y12 receptor is involved in expanding microglial processes by being stimulated by ADP, and these processes extend their projections toward the injury site. P2Y12 expression is microglial phenotype-dependent because it is highly expressed in anti-inflammatory M2 microglia but less in activated M1 microglia [36], suggesting that the P2Y12 receptor plays an essential role in microglial polarization.
Stimulating A2a receptors increases extracellular toxic glutamate release from their respective nerve terminals, activating microglial cells. A2a receptor activation also upregulates M1 microglial markers, but A2a receptor antagonists considerably reduce inflammatory cytokine gene expression [37]. A2a receptors are also involved in microglial morphological changes. P2Y12 receptor expression, which functions in microglial process expansion, is rapidly downregulated upon microglial activation, whereas concomitant A2a receptor upregulation mediates process regression in activated microglial cells [38]. Furthermore, subsequent changes in the expressed receptors cause a transition to the amoeboid microglial morphology characteristic during CNS inflammation (Figure 2).
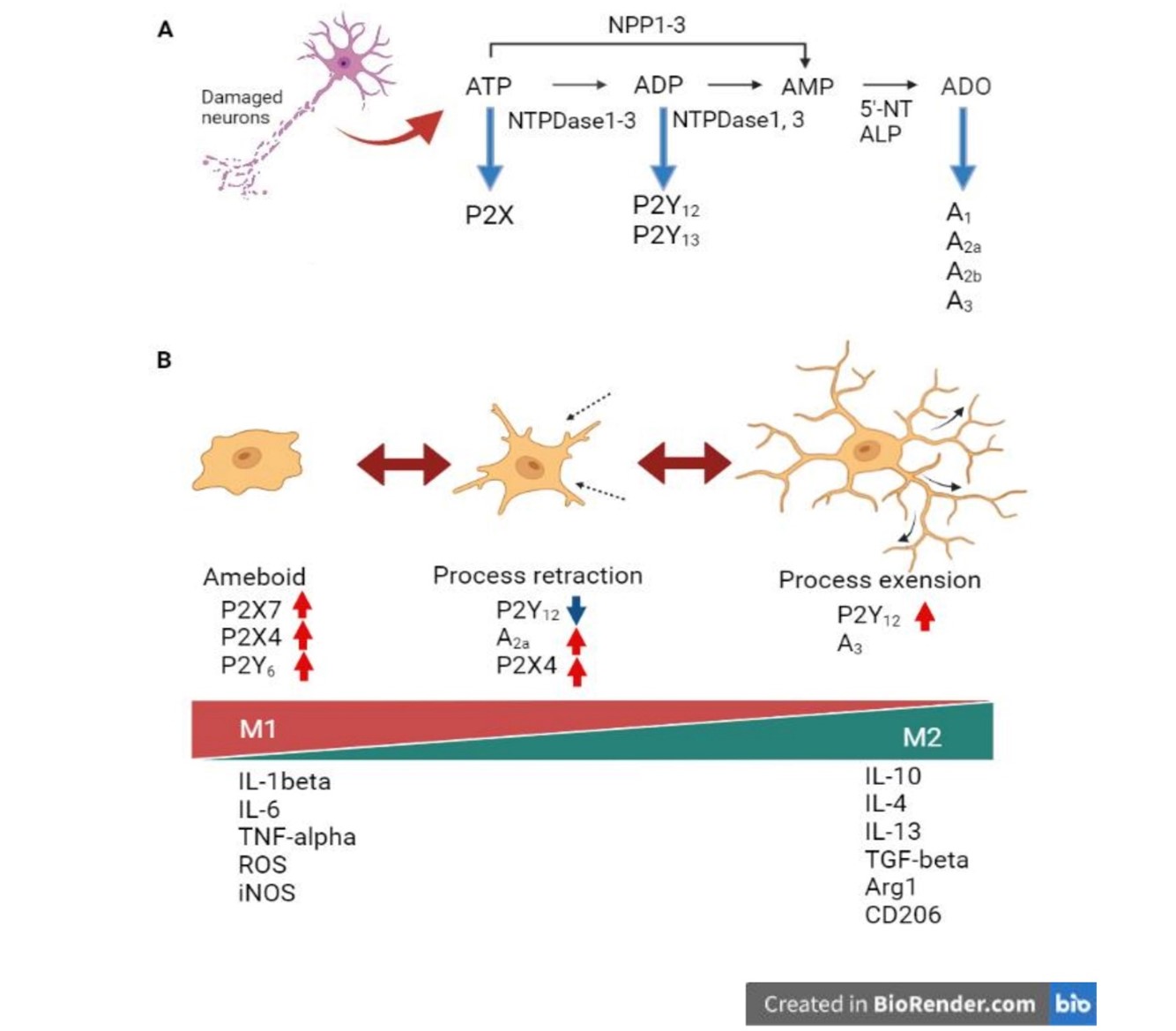
Figure 2. Activated states of microglia and expression of purinergic receptors. A. Ectonucleotidase ATP dephosphorylation. ATP is hydrolyzed to AMP by NPDases and NPPs; AMP is degraded to adenine (ADO) by 5'-NT and ALP. Arrows indicate the receptors for each nucleotide and nucleoside. B. Activated states of microglia and expression of purinergic receptors. ATP released extracellularly from damaged neurons first upregulates microglial P2Y12 and A3 receptors [39], and P2Y12 receptor stimulation extends the process toward the injured site. These processes are retracted by downregulating the P2Y12 receptor and upregulating the A2a receptor, altering the amoeboid phenotype. In amoeboid microglia, the expression of P2X7, P2Y4, and P2Y6 receptors is upregulated. P2X7 receptor is a major driver of inflammation and releases pro-inflammatory cytokines, chemokines, reactive oxygen or nitrogen species, growth factors, and excitotoxic glutamate [40]. P2X4 receptor may also be involved in neuronal excitotoxic damage [41]. The highest agonist at the P2Y6 receptor is UDP. UDP binding to the P2Y6 receptor induces microglial phagocytosis [42]. Ameboid microglia exhibit the inflammatory M1 phenotype, while microglia expressing the P2Y12 receptor exhibit the anti-inflammatory M2 phenotype [36], and express markers associated with each activation phenotype. This figure is drawn by Bio Render.com.
The alteration of purinergic receptor expression in TSC2 cKO mice by sirolimus treatment
We investigated the expression of P1 and P2 receptors on microglia because the microglia morphology in TSC2 cKO mice was significantly altered by sirolimus treatment (Figures 1A and 1B) [16]. In Figure 1C, color intensity depicts the expression levels of receptors and markers, with darker red and blue indicating higher and lower expression, respectively. TSC2 cKO mice had low expression of the P2Y12 receptor and high expression of the A2a receptor. The expression of M1 microglial markers, IL-6 and IL-1β, was insignificantly high in TSC2 cKO mice (Figure 1C); however, A2a receptor expression, which activates microglial cells, was significantly increased. This indicates that microglia in TSC2 cKO mice are in an incomplete M1 phenotypic state but may be intermediates between the M1 and M2 states (Figures 2 and 3). Furthermore, sirolimus treatment of TSC2 cKO mice increased the expression of the ADP-sensitive P2Y12 receptor (Figure1C). Expression of the P2Y12 receptor in microglia is highly expressed in anti-inflammatory M2 microglia but poorly expressed in activated M1 microglia [37]. M2 phenotypic markers were also upregulated in the microglia of sirolimus-treated TSC2 cKO mice (Figure 1C), suggesting that sirolimus treatment can shift microglia toward the M2 phenotype.
Ectonucleotidases
Ectonucleotidases are enzymes that hydrolyze extracellular ATP released from damaged cells and regulate various tissue functions. For example, nucleoside triphosphate diphosphohydrolase (NTPDase), an enzyme that hydrolyzes extracellular ATP and ADP, plays a major role in the hydrolysis of cell surface-located nucleotides (Figure 2A). Other enzymes that function in nucleotide hydrolysis include ectonucleotide pyrophosphatase phosphodiesterase and alkaline phosphatase [40]. Ectonucleotidase NTPDase1 hydrolyzes ATP and ADP to adenosine monophosphate, and 5′-nucleotidase degrades adenosine monophosphate to adenosine. In contrast, NTPDase2 degrades ATP but little ADP. Among the purinergic receptors, the P2X receptor binds to ATP while the P2Y12 receptor binds to ADP; thus, ectonucleotidase expression affects the response of these receptors (Figure 2A).
Ectonucleotidases expression in TSC2 cKO mice
Sirolimus treatment of TSC2 cKO mice increased NTPDase2 expression, promoting ATP hydrolysis to ADP (Figure 1C). This is consistent with the fact that sirolimus treatment increased the expression of ADP-sensitive P2Y12 receptors. Applying these results to previous findings (Figure 2), the microglia of TSC2 cKO mice did not have the morphology of ameboid-type microglia. However, the expression of A2a receptors that activate microglial cells increased. However, the M1 marker IL-6 was insignificantly elevated, nor were P2X4 and P2X7, which are involved in the expression of pro-inflammatory cytokines, suggesting that microglia are in an intermediate state between M1 and M2, rather than strictly M1 (Figure 3). On the other hand, sirolimus treatment increased the expression of NTPDase2 and P2Y12 receptors, suggesting that sirolimus polarizes microglia to the M2 phenotype.
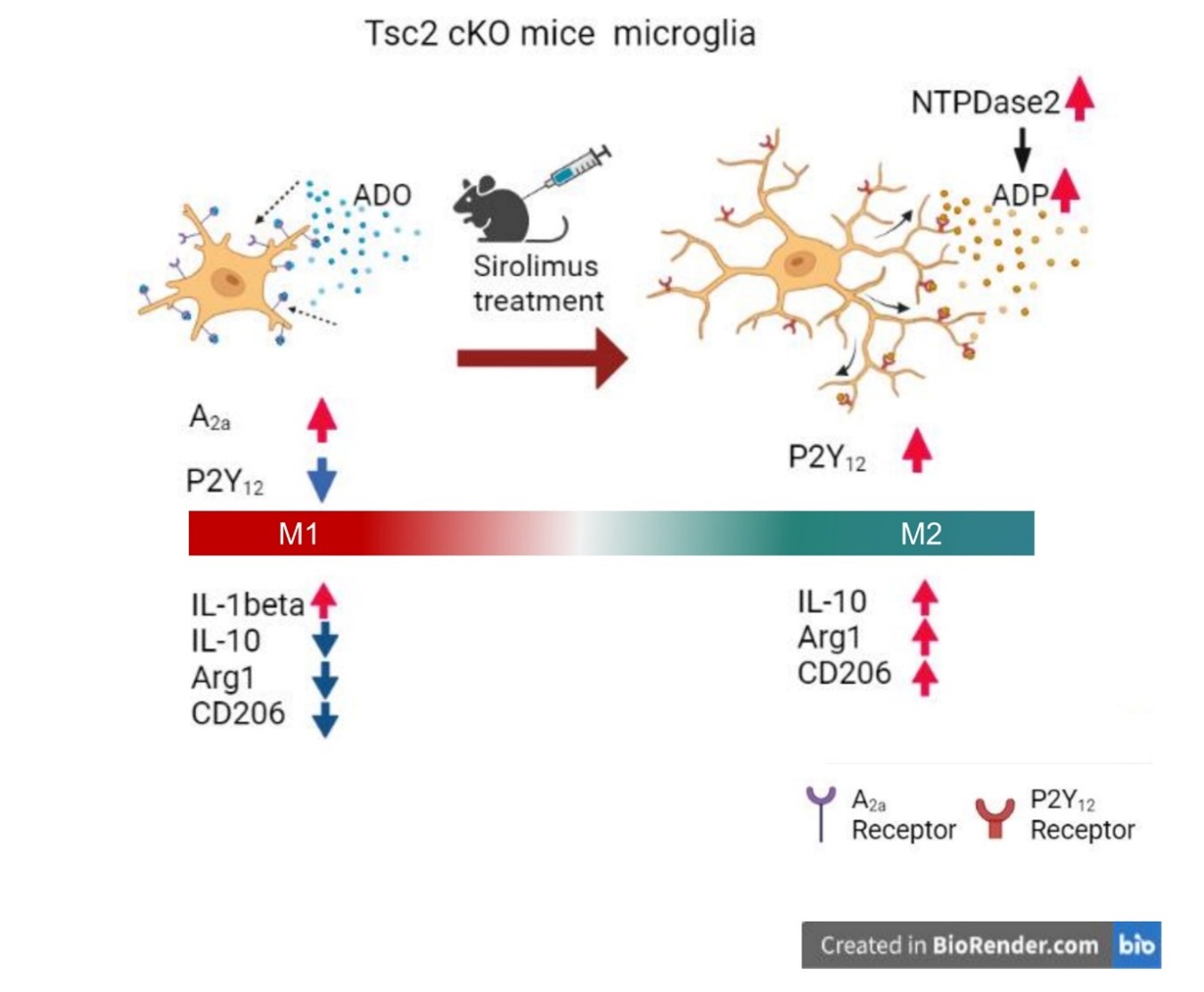
Figure 3. Microglial changes in sirolimus-treated TSC2 cKO mice. NTPDase2 is upregulated in the microglia of sirolimus-treated TSC2 cKO mice, which increases ADP. Furthermore, stimulating ADP via P2Y12 receptors extends the process. The microglia of TSC2 cKO mice without sirolimus treatment have upregulated A2a receptors. However, their microglia are between the M1 and M2 phenotypes rather than a strict M1 phenotype. Sirolimus treatment increases the expression of M2 phenotypic markers, suggesting that sirolimus-induced microglia shifted to the M2 phenotype.
Conclusion
TSC2 cKO mice exhibited epilepsy and TAND symptoms, which were treated with sirolimus. Furthermore, sirolimus treatment in TSC2 cKO mice shifted microglia to an M2 phenotype.
What factors contribute to the shift of microglia to the M2 phenotype by sirolimus treatment?
In TSC, mTORC1 hyperactivity is one of the etiological factors; however, activating mTORC1 inhibits autophagy. In mammalian cells, rapamycin (sirolimus) treatment alone has been shown to induce autophagy sufficiently [43]. In addition, autophagy upregulation promotes the polarization of microglia towards the M2 phenotype [44,45]. The polarization of microglia to the M2 phenotype in TSC2 cKO mice by sirolimus treatment suggests the possibility of increased autophagy due to mTORC1 inhibition.
Microglial inflammation due to M1 polarization is a common exacerbating factor in neurodegenerative diseases. Moreover, shifting microglial polarization to an M2 phenotype may be an effective strategy in treating neuroinflammatory diseases. For example, the natural carotenoid Astaxanthin promoted M2 polarization and suppressed neuroinflammation [46]. Furthermore, sirolimus, which shifts microglia to anti-inflammatory properties, may be a potential treatment for epilepsy and TAND in TSC and other neurodegenerative diseases.
Conflicts of interest
The authors declare no conflicts of interest.
Funding
This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (JP21K08322), from the Ministry of Health, Labour, and Welfare of Japan (20FC1043), and from a practical research project for rare and intractable diseases from the Japan Agency for Medical Research and Development (AMED) (20ek0109347h0003, 21lm0203007j0005, 22ym0126809j0001).
Acknowledgments
We thank the staff at the Center for Medical Research and Education, Graduate School of Medicine, the CoMIT Omics Center, the Institute of Experimental Animal Sciences, Graduate School of Medicine and the laboratory assistants at the Department of Dermatology at Osaka University for technical support and establishing animals and breeding. We would like to thank Editage (www.editage.com) for English language editing.
Author Contributions Statement
Dr. Koike-Kumagai contributed to the research and writing of this article; Dr. Wataya-Kaneda contributed to the editing and supervision of this review.
References
2. De Vries PJ, Wilde L, de Vries MC, Moavero R, Pearson DA, Curatolo P. A clinical update on tuberous sclerosis complex-associated neuropsychiatric disorders (TAND). American Journal of Medical Genetics Part C Seminars in Medical Genetics. 2018 Sep;178(3):309-320.
3. Gipson TT, Johnston M v. New insights into the pathogenesis and prevention of tuberous sclerosis-associated neuropsychiatric disorders (TAND). F1000Research. 2017 Jun 9;6. F1000 Faculty Rev-859.
4. Muzykewicz DA, Newberry P, Danforth N, Halpern EF, Thiele EA. Psychiatric comorbid conditions in a clinic population of 241 patients with tuberous sclerosis complex. Epilepsy & Behavior: E&B. 2007 Dec 1;1(4):506-513.
5. de Vries PJ, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. TSC-associated neuropsychiatric disorders (TAND): findings from the TOSCA natural history study. Orphanet Journal of Rare Diseases. 2018 Sep 10;13(1):157.
6. Ridler K, Suckling J, Higgins NJ, de Vries PJ, Stephenson CME, Bolton PF, et al. Neuroanatomical correlates of memory deficits in tuberous sclerosis complex. Cerebral cortex (New York, NY: 1991). 2007 Feb 17;17(2):261-271.
7. Orlova KA, Crino PB. The tuberous sclerosis complex. Annals of the New York Academy of Sciences. 2010 Jan; 1184:87-105.
8. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. The New England Journal of Medicine. 2010 Nov 4;363(19):1801-1811.
9. Koenig MK, Bell CS, Hebert AA, Roberson J, Samuels JA, Slopis JM, et al. Efficacy and Safety of Topical Rapamycin in Patients with Facial Angiofibromas Secondary to Tuberous Sclerosis Complex: The TREATMENT Randomized Clinical Trial. JAMA Dermatology. 2018 Jul 1;154(7):773-780.
10. Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Annals of Neurology. 2008 Apr;63(4):444-453.
11. Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/- mouse model of tuberous sclerosis. Nature Medicine. 2008 Aug;14(8): 843-848.
12. Reith RM, McKenna J, Wu H, Hashmi SS, Cho SH, Dash PK, et al. Loss of Tsc2 in Purkinje cells is associated with autistic-like behavior in a mouse model of tuberous sclerosis complex. Neurobiology of Disease. 2013 Aug; 51:93-103.
13. French JA, Lawson JA, Yapici Z, Ikeda H, Polster T, Nabbout R, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet (London, England). 2016 Oct 29;388(10056):2153-2163.
14. Krueger DA, Sadhwani A, Byars AW, de Vries PJ, Franz DN, Whittemore VH, et al. Everolimus for treatment of tuberous sclerosis complex-associated neuropsychiatric disorders. Annals of Clinical and Translational Neurology. 2017 Dec;4(12):877-887.
15. Overwater IE, Rietman AB, Mous SE, Bindels-De Heus K, Rizopoulos D, ten Hoopen LW, et al. A randomized controlled trial with everolimus for IQ and autism in tuberous sclerosis complex. Neurology. 2019 Jul 9; 93(2):E200-E209.
16. Koike-Kumagai M, Fujimoto M, Wataya-Kaneda M. Sirolimus relieves seizures and neuropsychiatric symptoms via changes of microglial polarity in tuberous sclerosis complex model mice. Neuropharmacology. 2022 Nov 1;218:109203.
17. Yang F, Yang L, Wataya-Kaneda M, Yoshimura T, Tanemura A, Katayama I. Uncoupling of ER/Mitochondrial Oxidative Stress in mTORC1 Hyperactivation-Associated Skin Hypopigmentation. Journal of Investigative Dermatology. 2018 Mar;138(3):669-678.
18. Yang F, Yang L, Wataya-Kaneda M, Teng L, Katayama I. Epilepsy in a melanocyte-lineage mTOR hyperactivation mouse model: A novel epilepsy model. PLOS ONE. 2020 Jan 24; 15(1):e0228204.
19. Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Translational neurodegeneration. 2020 Nov 26;9(1):2.
20. Sarejloo S, Khanzadeh S, Hosseini S, Gargari MK, Lucke-Wold B, Mosalamiaghili S, et al. Role of the Neutrophil to Lymphocyte Ratio in Guillain Barré Syndrome: A Systematic Review and Meta-Analysis. Mediators of Inflammation. 2022 Sep 12; 2022:3390831.
21. Hosseini S, Mofrad AME, Mokarian P, Nourigheimasi S, Azarhomayoun A, Khanzadeh S, et al. Neutrophil to Lymphocyte Ratio in Epilepsy: A Systematic Review. Mediators of Inflammation. 2022; 2022: 4973996.
22. Lyman M, Lloyd DG, Ji X, Vizcaychipi MP, Ma D. Neuroinflammation: the role and consequences. Neuroscience Research. 2014 Feb;79:1-12.
23. El Ali A, Rivest S. Microglia Ontology and Signaling. Frontiers in Cell and Developmental Biology. 2016 Jun 29;4:72.
24. Colonna M, Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annual Review of Immunology. 2017 Apr 26;35:441-468.
25. Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease - A double-edged sword. Neuron. 2002 Aug 1;35(3):419-432.
26. Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation Induces Neurodegeneration. Journal of Neurology, Neurosurgery and Spine. 2016;1(1):1003.
27. Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Human Molecular Genetics. 2009 Feb 1;18(3):482-496.
28. Guo S, Wang H, Yin Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Frontiers in Aging Neuroscience. 2022 Feb 16;14 :815347.
29. Rosi S. A polarizing view on posttraumatic brain injury inflammatory response. Brain Circulation. 2016 Jul-Sep;2(3):126-128.
30. Burnstock G. Physiology and pathophysiology of purinergic neurotransmission. Physiological Reviews. 2007 Apr;87(2):659-797.
31. Davalos D, Grutzendler J, Yang G, Kim J v., Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nature Neuroscience. 2005 Jun;8(6):752-758.
32. A. Weisman G, T. Woods L, Erb L, I. Seye C. P2Y receptors in the mammalian nervous system: pharmacology, ligands and therapeutic potential. CNS & Neurological Disorders Drug Targets. 2012 Sep;11(6):722-738.
33. Haynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan W-B, et al. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nature Neuroscience 2006 Dec;9(12):1512-1519.
34. Eyo UB, Peng J, Swiatkowski P, Mukherjee A, Bispo A, Wu L-J. Neuronal Hyperactivity Recruits Microglial Processes via Neuronal NMDA Receptors and Microglial P2Y12 Receptors after Status Epilepticus. The Journal of Neuroscience. 2014 Aug 6;34(32):10528-10540.
35. Lou N, Takano T, Pei Y, Xavier AL, Goldman SA, Nedergaard M. Purinergic receptor P2RY12-dependent microglial closure of the injured blood-brain barrier. Proceedings of the National Academy of Sciences of the United States of America. 2016 Jan 26;113(4):1074-1079.
36. Moore CS, Ase AR, Kinsara A, Rao VTS, Robinson MM, Leong SY, et al. P2Y12 expression and function in alternatively activated human microglia. Neurology(R) Neuroimmunology & Neuroinflammation. 2015 Mar 19;2(2):e80.
37. Colella M, Zinni M, Pansiot J, Cassanello M, Mairesse J, Ramenghi L, et al. Modulation of Microglial Activation by Adenosine A2a Receptor in Animal Models of Perinatal Brain Injury. Frontiers in Neurology. 2018 Sep 11; 9:605.
38. Orr AG, Orr AL, Li XJ, Gross RE, Traynelis SF. Adenosine A(2A) receptor mediates microglial process retraction. Nature Neuroscience. 2009 Jul;12(7):872-878.
39. Ohsawa K, Sanagi T, Nakamura Y, Suzuki E, Inoue K, Kohsaka S. Adenosine A3 receptor is involved in ADP-induced microglial process extension and migration. Journal of Neurochemistry. 2012 Apr;121(2):217-227.
40. Haas CB, Lovászi M, Braganhol E, Pacher P, Haskó G. Ectonucleotidases in Inflammation, Immunity, and Cancer. Journal of Immunology. 2021 May 1;206(9):1983-1990.
41. Ulmann L, Levavasseur F, Avignone E, Peyroutou R, Hirbec H, Audinat E, et al. Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia. 2013 Aug;61(8):1306-1319.
42. Koizumi S, Shigemoto-Mogami Y, Nasu-Tada K, Shinozaki Y, Ohsawa K, Tsuda M, et al. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature. 2007 Apr 26;446(7139):1091-1095.
43. Noda T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules. 2017 Sep;7(3):52.
44. Jin MM, Wang F, Qi D, Liu WW, Gu C, Mao CJ, et al. A Critical Role of Autophagy in Regulating Microglia Polarization in Neurodegeneration. Frontiers in Aging Neuroscience. 2018;10:378.
45. Bai J, Geng B, Wang X, Wang S, Yi Q, Tang Y, et al. Exercise Facilitates the M1-to-M2 Polarization of Microglia by Enhancing Autophagy via the BDNF/AKT/mTOR Pathway in Neuropathic Pain. Pain Physician. 2022 Oct;25(7):E1137-E115.
46. Wen X, Xiao L, Zhong Z, Wang L, Li Z, Pan X, et al. Astaxanthin acts via LRP-1 to inhibit inflammation and reverse lipopolysaccharide-induced M1/M2 polarization of microglial cells. Oncotarget. 2017 Sep 3;8(41):69370-69385.