Abstract
Methylphenidate (MPD) is a psychostimulant that is widely used to treat attention deficit hyperactivity disorder, and is being increasingly misused as a recreational drug and cognitive enhancer. MPD acts on the reward system of the CNS through specific signaling pathways to produce its effects on behavior, including tolerance, withdrawal, and sensitization. The nucleus accumbens (NAc) is one of the predominant components of this system, however the role of the NAc’s glutaminergic system in the behavioral response to MPD has not been studied. The objective of this study was to assess the role of glutaminergic signaling and the response to acute and chronic MPD exposure as measured by three different locomotive behaviors following selective bilateral lesions. Three groups of n=8 rats were used: control, sham NAc lesions, and glutaminergic-specific (ibotenic acid toxin) NAc lesions. On experimental day (ED) 1, all groups received saline injections to establish a baseline. On ED 2, NAc surgeries took place, followed by a 5-day recovery period (ED 3-7). On ED 8 saline was given to obtain a post-surgical baseline. Groups then received six daily MPD 2.5 mg/kg injections (ED 9-14) to produce a chronic effect, behavioral sensitization in this study, then three days of washout with no injection (ED 15-17), followed by a re-challenge with the previous 2.5 mg/kg MPD dose on ED 18. Locomotive activity was recorded for 60 minutes after each injection by a computerized animal activity monitoring system. All groups showed an increase in behavioral activity following acute MPD exposure, and developed behavioral sensitization following chronic MPD exposure that was maintained after three days of MPD washout. Compared to controls and sham lesions, the horizontal activity response was significantly (P<0.05) attenuated both acutely and chronically following glutaminergic selective ibotenic acid lesions to the NAc however the other indices showed no change. This indicates that glutaminergic signaling in the NAc plays a role in modulating the response to acute and chronic MPD.
Keywords
Methylphenidate, Ritalin, Nucleus accumbens, Lesion, Sensitization, Glutamate
Introduction
Methylphenidate (MPD), more commonly known at Ritalin® or Concerta®, is a psychostimulant that is prescribed to treat behavioral disorders such as attention deficit hyperactivity disorder (ADHD) but is increasingly being misused and abused as a cognitive performance enhancer or recreational stimulant in normal individuals [1-5]. This has been driven by the rapid increase in patients diagnosed with Attention Deficit Hyperactivity Disorder (ADHD) for which MPD is the drug of choice [6-10], as well as the rise in non-prescription use of MPD for academic enhancement and recreation [7,11-15].
This is of concern as MPD shares pharmacologic characteristics with other addictive psychostimulants such as amphetamine and cocaine, and could thus share similar addictive potential [6,16-20]. MPD, like amphetamine and cocaine, acts as an indirect dopamine agonist by inhibiting the dopamine reuptake at the presynaptic terminal, leading to increased dopamine within the synaptic cleft [21-23]. Acute MPD exposure produces an increase in behavioral locomotor activity; chronic use elicits sensitization, tolerance, and/or withdrawal which are behavioral markers indicating a substance has the potential to elicit dependence [17,24-29]. Sensitization is a sustained increase in behavioral activity beyond the acute effect following chronic administration of a substance [30,31].
The central nervous system’s (CNS) reward system is known to participate in the long-term changes associated with substance abuse [32-37]. The circuit consists of multiple CNS structures, however the core pathway is the mesolimbic pathway in which dopaminergic neurons from the ventral tegmental area (VTA) project to the nucleus accumbens (NAc) and the ventral striatum, then onwards to the prefrontal cortex (PFC). The Nucleus Accumbems (NAc) is a reward circuit structure that is critical for motivation, emotion, limbic functions, and motor execution [30,38-43]. Non-specific and dopaminergic specific lesions to the NAc have shown it to be critical to regulating the response to MPD [44,45], however the role of the glutaminergic system remains uninvestigated. Glutaminergic signaling has been shown to modulate the long-term response between other reward/motive circuit nuclei [26,28,29,36,44,46-60], and it known to participate in inputs to the NAc, however its role in the acute and chronic response to MPD is unknown.
This study set out to determine if the glutaminergic system of the NAc participates in the response to MPD. To do this, 3 groups of animals were used: NAc intact controls, sham lesions, and specific glutaminergic chemical lesions. Animals were exposed to acute and chronic (repetitive) MPD and the response was monitored with a computerized monitoring system in an open field assay.
Methods
Animals
Twenty-four male Sprague-Dawley rats weighing 170- 180g were obtained from Harlan Labs (Indianapolis, IN, USA). Animals were individually placed in plexiglass cages (40.5x40.5x31.5 cm in dimension) in a soundproof room without disturbance to the experimental environment for 4-5 days to acclimate prior to experimentation. These cages served as the home and test cage. Animals were maintained on a 12-hour light/dark cycle that began at 06:00. Food and water were provided ad libitum throughout the experiment, and the temperature was kept at 21 ± 2°C with a relative humidity of 37-42%. At the beginning of the experimental phase, the rats were weighed and randomly divided into three groups: NAc-intact controls (n=8), sham operation (n=8), and ibotenic acid chemical ablation of the glutaminergic system (n=8). This protocol was approved by our Animal Welfare Committee and carried out in accordance with the National Institute of Health Guide for Care and Use of Laboratory Animals.
Experimental procedure (Table 1)
Rats were given 4-5 days to acclimate in their home cage before experimentation. On experimental day 1 (ED 1-Sal) animals were weighed and 0.8 mL of 0.9% saline was administered intra-peritoneal (ip). All animals weighed 200-220 g at that time. Locomotive behavioral activity was recorded for 120 minutes post-injection to establish a baseline prior to surgical manipulation. On experimental day 2 (ED 2), the lesion and sham groups underwent surgery and were then allowed to recover for approximately 5 days (ED 3-7). On experimental day 8, saline was re-administered (ED 8-Sal) and postsurgical locomotor activity was recorded for 120 minutes to compare with the pre-surgical baseline (ED 1-Sal). Starting on experimental day 9 (ED 9-MPD), daily injections of 2.5 mg/kg MPD (Mallinckrodt, Hazelwood MO) dissolved in 0.8 mL of 0.9% saline were administer for 6 consecutive days (ED 9-MPD to ED 14-MPD), and activity recorded for 120 minutes post-injection. This dose of 2.5 mg/kg MPD has been shown to be sufficient to elicit behavioral sensitization in rats in previous doseresponse experiments [27-29,48,51,61-68]. For the next 3 days (ED 15-17), animals received no injections (the washout period). After the washout period (ED 18-MPD), the rats were re-challenged with MPD at the previous dose of 2.5 mg/kg and behavioral activity was observed for 60 minutes (the expression phase). All boluses were given at approximately 07:30 in the morning in 0.8 mL volumes.
Surgical Procedure (ED 2)
On ED 2, the sham operation group, the electrolytic lesion group, the 6-OHDA group, and the ibotenic acid group animals were anaesthetized with 60 mg/ kg pentobarbital and placed in the stereotactic apparatus. An incision was to expose the skull. For surgery, holes were drilled in the skull 1.7 mm anterior from the bregma and 1.6 mm lateral to the midline bilaterally based on the co-ordinates derived from Paximos and Watson Rat Brain Atlas [69].
Sham operation: For the sham group, the animal was anesthetized, the skin opened, holes drilled in the skull, and a 27G cannula was inserted bilaterally to a depth of 6.8 mm but no agent administered. The cannulas were then removed, and the incision closed with wound staples.
NAc Glutaminergic system ablation: For the glutaminergic ablation group, ibotenic acid, a glutaminergic toxin, was employed [70-74]. A 27G cannula was inserted bilaterally to a depth of 6.8 mm. 5 μg of ibotenic acid was dissolved in 5 μl of 0.9% normal saline was slowly infused then the cannula left in place for 6 minutes to allow for full diffusion. The cannulas were then removed, and the incision closed with wound staples.
Apparatus
Behavioral locomotive activity was recorded using the open field computerized animal activity monitoring system (CAAM, AccuScan Instruments, Inc., Columbus OH). The CAAM system consists of 2 arrays of 16 infrared light beams with sensors on the opposite side, spaced every 2.5 cm that cross orthogonally through the plexiglass cage. Sensor polling frequency was set at 100 Hz. Movement of the rats interrupted the infrared light beams, and each beam-break detected by a sensor was collected as an event by the AccuScan Analyzer and transferred to a computer. Events over a 5-minute period were summed, giving 12 5-minute bins for each hour of observation. These bins were transferred to the OASIS data collecting software and three indices of behavioral locomotion were compiled for each collection period: total travelling distance (TD)- all forward locomotion in cm, horizontal activity (HA)- the overall movement in the lower level of the cage, and the number of stereotypic movements (NOS)- episodes of purposeless, repetitive movement in the upper level of the sensors separated by at least 1 second.
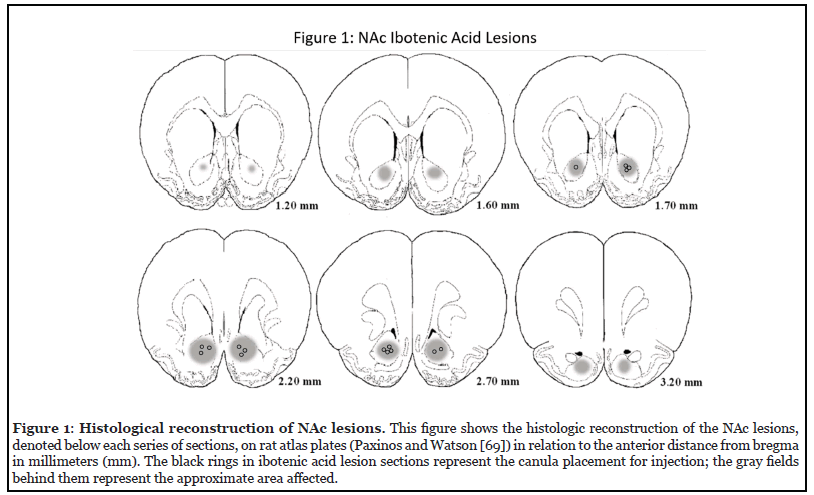
Histology (Figure 1)
At the conclusion of the experiment, animals were overdosed with sodium pentobarbital and perfused with 10% formaldehyde. The brains were removed stored in 10% formaldehyde. 60 μm thickness corona l sections were cut, stained, and scanned with a high-resolution scanner to identify lesion size and location correlated to the NAc using the Paximos and Watson rat brain atlas [69] (Figure 1).
Data analysis
Rat behavioral locomotive activity was quantified by three compiled indices of movement (HA, TD, NOS) obtained in twelve 5-minute bins collected the hour after injections for each rat were averaged across each experimental group based on the experimental day to allow for comparisons. Post-surgical manipulation effects on baseline behavioral locomotor activity were determined by comparing the animal’s activity after a saline injection before and after the surgical intervention (ED 8-Sal vs. ED 1-Sal). The acute effects of MPD were determined by comparing the first day of MPD administration to the post-surgical baseline (ED 9-MPD vs. ED 8-Sal). The effects of repetitive (chronic) MPD exposure over 6 consecutive days on behavioral locomotor activity were determined by comparing the final day of administration to the first, i.e. the induction phase (ED 14-MPD vs. ED 9-MPD). The effects of chronic MPD exposure following a washout period on behavioral locomotor activity were determined by comparing MPD re-challenge to the initial administration, i.e the expression phase (ED 18-MPD vs. ED 9-MPD) (See Table 1). Significance of change among these withingroup comparisons was determined by ANOVA, with repeated measures with adjustments for correlation among measurements within each animal. Post ad hoc comparisons were used to estimate changes between days within groups. A p-value<0.05 was considered statistically significant. The effects of the ibotenic acid lesion were determined by comparing the ibotenic acid lesion group to both the control and sham groups on each of the recording days (ED 1-Sal, ED 8-Sal, ED 9-MPD, ED 14-MPD, and ED 18-MPD). Significance of change among the betweengroup comparisons was determined with Turkey-Kramer Honest Significant Difference (HSD) post hoc test. A p-value<0.05 was considered statistically significant.
| Group | Experimental Schedule | ||||||
| ED 1* | ED 2 | ED 3-7 | ED 8* | ED 9-14* | ED 15-17 | ED 18* | |
| Control | Saline | Saline | MPD | Washout | MPD re- challenge | ||
| Sham | Saline | Surgery | Recovery | Saline | MPD | Washout | MPD re- challenge |
| Ibotenic acid lesion | Saline | Surgery | Recovery | Saline | MPD | Washout | MPD re- challenge |
Table 1: Methylphenidate administration schedule. The table shows the experimental treatment protocol for the 5 groups of rats used. Each group consisted of N=8 rats. Displayed are the experimental days (ED’s) either normal saline or methylphenidate (MPD) 2.5 mg/kg ip was administered according to injection protocol, in a standardized volume of 0.8 ml at 07:30. * indicates day rats were behaviorally recorded post-injection. The experiment lasted 18 experimental days. The experimental schedule began after several days of acclimatization of the rats to their home/experimental cages.
Results
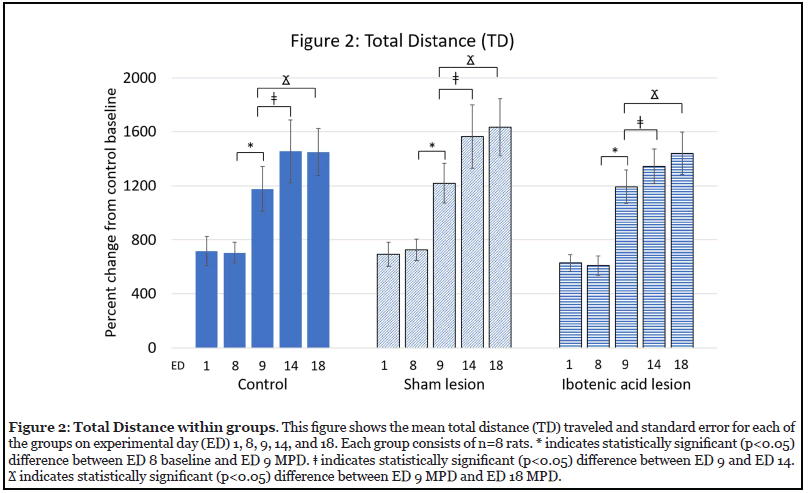
Effect of MPD on activity (Figure 2)
Figure 2 shows the effect of the MPD administration on total distance (TD) traveled on the five recording days (ED 1-Sal, ED 8-Sal, ED 9-MPD, ED 14-MPD, and ED 18-MPD) for the NAc control, sham, and ibotenic acid lesion groups. Surgery with or without chemical intervention to the NAc (ED 8-Sal vs. ED 1-Sal) did not lead to a statistically significant change in TD for the sham and ibotenic acid lesion groups as compared to the control group (Figure 2). Similar results were seen in horizontal activity (HA) and number of stereotypic movements (NOS). This observation indicates that animal handling, injection volume, and injection procedure were cons istent, and that the surgical intervention did not modulate baseline activity. The administration of 2.5 mg/kg MPD yielded a statistically significant (* p<0.05) increase in TD following MPD exposure for all groups relative to their postsurgical baseline (ED 9-MPD vs. ED 8-Sal) (Figure 2). Similar results were seen in HA and NOS. Administration of a repetitive 2.5 mg/kg MPD dose for an additional five consecutive days resulted in a further statistically significant (? p<0.05) increase in TD beyond the acute effect of MPD for all groups (ED 14-MPD vs. ED 9-MPD) (Figure 2). Similar results were seen in HA and NOS. This further augmentation in locomotive behavior following repeated exposure to MPD confirms that 2.5 mg/kg MPD induces behavioral sensitization. Re-challenge with the same 2.5 mg/kg MPD dose after a three-day washout period following chronic MPD exposure (six days of MPD administration) caused all groups to again show a further statistically significant (? p<0.05) increase in TD as compared to acute MPD administration (ED 18-MPD vs. ED 14-MPD) (Figure 2). Similar results were seen in HA and NOS. This continued augmentation of the response to MPD even after drug washout is the continued expression of sensitization to chronic psychostimulant use, i.e. the expression phase.
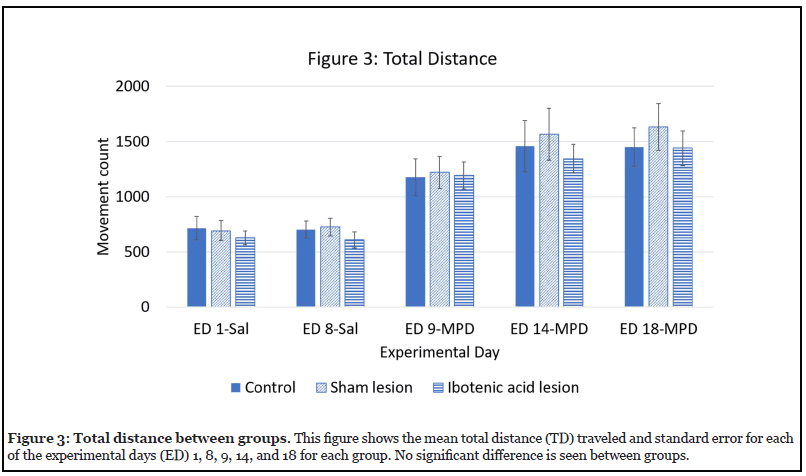
Effect of ibotenic acid lesion on TD (Figure 3)
Figure 3 shows the effects of ibotenic acid lesions to the NAc on TD by comparing each group (control, sham, and ibotenic acid lesion) to the other two groups on each experimental day. No significant difference was seen between any of the groups on any of the experimental days, i.e. all groups behaved similarly in response to MPD exposure.
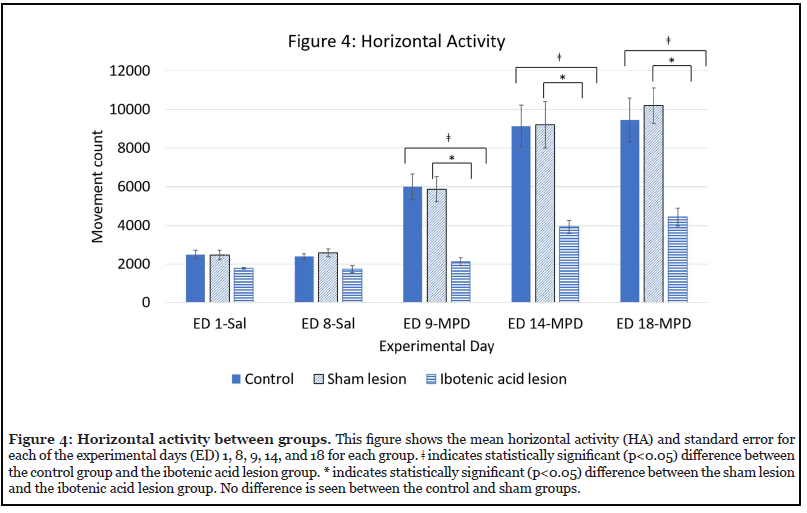
Effect of ibotenic acid lesion on HA (Figure 4)
Figure 4 shows the effects of ibotenic acid lesions to the NAc on HA by comparing each group (control, sham, and ibotenic acid lesion) to the other two groups on each experimental day. Compared to the control and sham groups, the group that received lesions to the NAc showed a significant difference between the control (? p<0.05) and the sham (* p<0.05) groups in response to MPD both acutely (ED 9-MPD) and chronically (ED 14-MPD and ED 18-MPD).
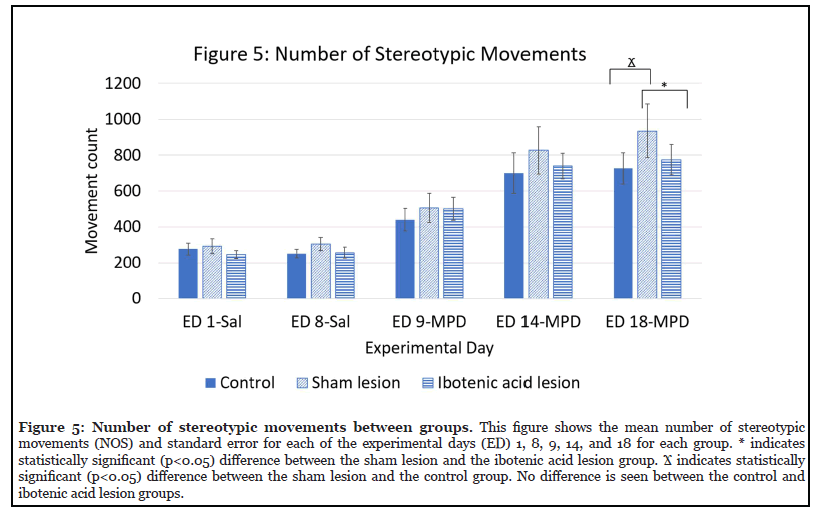
Effect of ibotenic acid lesion on NOS (Figure 5)
Figure 5 shows the effects of ibotenic acid lesions to the NAc on NOS by comparing each group (control, sham, and ibotenic acid lesion) to the other two groups on each experimental day. No significant difference was seen between any of the groups on ED 9-MPD or ED 14-MPD. On ED 18-MPD, a significant difference was seen between the sham lesion group and both the NAc intact control (? p<0.05) and the ibotenic acid lesion groups (* p<0.05). No significant difference was seen between the control and ibotenic acid lesion group.
Discussion
This experiment was conducted to determine the role of glutaminergic signaling in the nucleus accumbens (NAc) in the response to acute and chronic methylphenidate (MPD). The findings of this work show that in NAc intact animals, 2.5 mg/kg MPD results in an acute increase in activity in all locomotor indices studied (TD, HA, NOS, Figure 2), and that chronic repetitive exposure results in behavioral sensitization- the further significant increase above the acute effect (Figure 2). This effect is clearly modulated following a specific bilateral glutaminergic lesion to the NAc with ibotenic acid, with HA specifically showing a consistent significant difference from the NAc intact control and sham groups following both acute and chronic 2.5mg/kg MPD exposure (Figure 4). This difference was absent in the other indices (TD and NOS, Figures 3 and 5 respectively). The sham lesion alone shows a difference from the control and ibotenic lesion groups, which appears to be a statistical artifact as the magnitude is similar to the NAc intact controls and the ibotenic acid lesion group and the difference is not seen on ED 14-MPD when the chronic effect of MPD is also manifested.
The NAc is a structure located near the anterior commissure that is critical for the motivation and rewardseeking behavior. It is composed primarily of dopaminergic medium spiny neurons (MSN’s) and is divided into a shell and a core that mediate different functions [75-80]. The NAc receives input primarily from the VTA, in addition to inputs from the substantia nigra, the amygdala, the hippocampus, and the PFC. The NAc outputs ascend to various basal ganglia and midbrain structures including the substantia nigra, the VTA, the ventral pallidum, the thalamus, the subpallidus, and the stria terminalis [78,81-84].
Previously reported lesions to the NAc have confirmed its role in mediating the behavioral response to MPD. Psychostimulants such as MPD cause an increase in dopaminergic transmission from the VTA to the NAc, and increased dopamine within the NAc leads to increased locomotion [85-87]. Direct chronic microinjection of dopaminergic agonists such as amphetamine, cocaine, or morphine into the NAc can induce sensitization [38,88-96], suggesting that the NAc is involved in the induction of behavioral sensitization. Non-specific lesions to the NAc have been shown to lead to an enhanced acute effect of MPD, but absent long-term behavioral changes such as sensitization following chronic exposure [44]. This this is also seen with amphetamine, cocaine, and nicotine [97-103]. Dopaminergic lesions to the NAc have produced more complex behavioral changes, with some animals exhibiting no increase in locomotor activity acute MPD exposure and others showing a significantly elevated locomotor activity following MPD exposure [45]. Animals that responded to MPD acutely did not develop behavioral sensitization, while those that showed no behavioral change following the dopaminergic lesion did show behavioral sensitization [45]. This work was noted to not determine lesion accuracy which could explain the dichotomy of animal responses, however it still indicated that accumbal dopaminergic signaling is critical for the response to psychostimulants.
Glutaminergic signaling in the NAc has been unexplored till this present study, but has been shown to be critical in other reward circuit nuclei [26,28,29,36,44,46-60]. This study found that following specific glutaminergic ablation of the NAc by ibotenic acid, animals showed the same general response to acute and chronic MPD exposure as the control and sham NAc lesion groups, with an acute increase in behavioral activity following MPD and then further augmentation with chronic exposure (Figure 2). However, when the different behavioral expressions (HA, TD, NOS) to MPD exposure were compared between groups, a significant attenuation in the behavioral activity comprising forward motion as measured by HA was seen following glutaminergic-specific lesions to the NAc (Figures 3-5). This attenuation of HA indicates that glutaminergic signaling in the NAc is critical in modulating behavior and influences signaling pathways differently. This fits with the current knowledge that glutaminergic inputs to the NAc come from other reward circuit nuclei [104,105], and with other work showing that glutaminergic signaling is responsible for modulating the core effect of MPD at other reward circuit nuclei [26,28,29,36,44,46-60]. It also seems to indicate that different subcortical circuits govern different behavioral responses, as animals with glutaminergic lesions to the NAc, HA exhibited significantly less behavioral activity in response to both acute and chronic MPD exposure. HA is a measure of forward motion, and can be regarded as a goal-directed behavior (although in this experiment the open field assay is not designed to stimulate this mode of activity) which would seem to imply that glutaminergic signaling specifically modulates motivational circuits versus generalized motor modulation, which would involve both TD and NOS.
Previous work initially determined the NAc shell to be critical for the excitatory response to psychostimulants, as it showed the greatest response in response to their administration [90,95,106,107]. However, it is increasingly being recognized that the NAc core also participates in the response to psychostimulants [108-111], and that both play a role in motivation and behavioral actions [112-114]. The results seen here agree with emerging work showing that while the NAc core and shell are anatomically distinct, distinct circuits between them govern different behavioral responses [108-114]. Targeting a spherical shell structure with a chemical lesion presents a substantial technical problem and further interrogation of these distinctions will require further work. The strength of the paper is the using of local injection of specific neurotoxin to eliminate the glutaminergic system in the NAc. Additional experiments are needed to use specific neurotoxin to study other neurotransmitters signaling.
Conclusion
The nucleus accumbens (NAc) is a rewards circuit structure that is critical for the response to MPD. It is divided into a shell and core that serve distinct roles in the response to psychostimulants such as MPD. Three different locomotive behaviors were studied, and it was found that lesions to the glutaminergic signaling pathways of the NAc result in significant attenuation of forward motion HA compared to control and sham groups. This difference was not seen in the other behaviors (TD and NOS), indicating that different NAc circuits govern specific behavioral expressions to acute and chronic MPD.
Acknowledgements
This study was supported by a National Institute of Health grant (NIH DA-027220).
References
2. Frauger E, Amaslidou D, Spadari M, Allaria- Lapierre V, Braunstein D, Sciortino V, et al. Patterns of methylphenidate use and assessment of its abuse among the general population and individuals with drug dependence. European Addiction Research. 2016;22(3):119-26.
3. King SA, Casavant MJ, Spiller HA, Hodges NL, Chounthirath T, Smith GA. Pediatric ADHD medication exposures reported to US poison control centers. Pediatrics. 2018 Jun;141(6):e20173872.
4. Pauly V, Frauger E, Lepelley M, Mallaret M, Boucherie Q, Micallef J. Patterns and profiles of methylphenidate use both in children and adults. British Journal of Clinical Pharmacology. 2018 Jun;84(6):1215-27.
5. Visser SN, Danielson ML, Bitsko RH, Holbrook JR, Kogan MD, Ghandour RM, et al. Trends in the parent- report of health care provider-diagnosed and medicated attention-deficit/hyperactivity disorder: United States, 2003–2011. Journal of the American Academy of Child & Adolescent Psychiatry. 2014 Jan 1;53(1):34-46.
6. Accardo P, Blondis TA. What’s all the fuss about Ritalin?. The Journal of Pediatrics. 2001 Jan 1;138(1):6-9.
7. Fond G, Gavaret M, Vidal C, Brunel L, Riveline JP, Micoulaud-Franchi JA, et al. (Mis) use of prescribed stimulants in the medical student community: motives and behaviors: a population-based cross-sectional study. Medicine. 2016 Apr;95(16):e3366.
8. Froehlich TE, Lanphear BP, Epstein JN, Barbaresi WJ, Katusic SK, Kahn RS. Prevalence, recognition, and treatment of attention-deficit/hyperactivity disorder in a national sample of US children. Archives of Pediatrics & Adolescent Medicine. 2007 Sep 1;161(9):857-64.
9. Stanford C, Tannock R (eds). Behavioral neuroscience of attention deficit hyperactivity disorder and its treatment. Springer Science & Business Media; 2012 Jan 10.
10. Storebø OJ, Simonsen E, Gluud C. Methylphenidate for attention-deficit/hyperactivity disorder in children and adolescents. Jama. 2016 May 10;315(18):2009-10.
11. Bogle KE, Smith BH. Illicit methylphenidate use: a review of prevalence, availability, pharmacology, and consequences. Current Drug Abuse Reviews. 2009 May 1;2(2):157-76.
12. Cox ER, Motheral BR, Henderson RR, Mager D. Geographic variation in the prevalence of stimulant medication use among children 5 to 14 years old: results from a commercially insured US sample. Pediatrics. 2003 Feb 1;111(2):237-43.
13. Djezzar S, Courné MA, Richard N. EPA-0748– Methylphenidate abuse: results of a french survey. European Psychiatry. 2014;29(S1):1.
14. Greely H, Sahakian B, Harris J, Kessler RC, Gazzaniga M, Campbell P, Farah MJ. Towards responsible use of cognitive-enhancing drugs by the healthy. Nature. 2008 Dec;456(7223):702-5.
15. Wilens TE, Adler LA, Adams J, Sgambati S, Rotrosen J, Sawtelle R, et al. Misuse and diversion of stimulants prescribed for ADHD: a systematic review of the literature. Journal of the American Academy of Child & Adolescent Psychiatry. 2008 Jan 1;47(1):21-31.
16. Diaz Heijtz R, Scott L, Forssberg H. Alteration of dopamine D1 receptor-mediated motor inhibition and stimulation during development in rats is associated with distinct patterns of c-fos mRNA expression in the frontalstriatal circuitry. European Journal of Neuroscience. 2004 Feb;19(4):945-56.
17. Kuczenski R, Segal DS. Effects of methylphenidate on extracellular dopamine, serotonin, and norepinephrine:comparison with amphetamine. Journal of Neurochemistry. 1997 May;68(5):2032-7.
18. Kharas N, Yang P, Castro-Alvarado D, Rose K, Dafny N. Exposure to methylphenidate in adolescence and adulthood modulates cross-sensitization to amphetamine in adulthood in three genetically variant female rat strains. Behavioural Brain Research. 2019 Apr 19;362:36-45.
19. Teo SK, Stirling DI, Hoberman AM, Christian MS, Thomas SD, Khetani VD. D-methylphenidate and D, L-methylphenidate are not developmental toxicants in rats and rabbits. Birth Defects Research Part B: Developmental and Reproductive Toxicology. 2003 Apr;68(2):162-71.
20. Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley JS, et al. Is methylphenidate like cocaine?: Studies on their pharmacokinetics and distribution in the human brain. Archives of General Psychiatry. 1995 Jun 1;52(6):456-63.
21. Gatley SJ, Volkow ND, Gifford AN, Fowler JS, Dewey SL, Ding YS, et al. Dopamine-transporter occupancy after intravenous doses of cocaine and methylphenidate in mice and humans. Psychopharmacology. 1999 Sep;146(1):93- 100.
22. John CE, Jones SR. Voltammetric characterization of the effect of monoamine uptake inhibitors and releasers on dopamine and serotonin uptake in mouse caudateputamen and substantia nigra slices. Neuropharmacology. 2007 Jun 1;52(8):1596-605.
23. Volkow ND, Wang GJ, Fowler JS, Fischman M, Foltin R, Abumrad NN, et al., Methylphenidate and cocaine have a similar in vivo potency to block dopamine transporters in the human brain. Life Sciences. 1999 May 28;65(1):PL7- 12.
24. Algahim MF, Yang PB, Wilcox VT, Burau KD, Swann AC, Dafny N. Prolonged methylphenidate treatment alters the behavioral diurnal activity pattern of adult male Sprague-Dawley rats. Pharmacology Biochemistry and Behavior. 2009 Mar 1;92(1):93-9.
25. Gaytan O, Sripada S, Swann A, Dafny N. Blockade of sensitization to methylphenidate by MK-801: partial dissociation from motor effects. Neuropharmacology. 2001 Jan 1;40(2):298-309.
26. Lee MJ, Swann AC, Dafny N. Methylphenidate sensitization is prevented by prefrontal cortex lesion. Brain Research Bulletin. 2008 May 15;76(1-2):131-40.
27. Yang PB, Amini B, Swann AC, Dafny N. Strain differences in the behavioral responses of male rats to chronically administered methylphenidate. Brain Research. 2003a May 9;971(2):139-52.
28. Yang PB, Swann AC, Dafny N. Sensory-evoked potentials recordings from the ventral tegmental area, nucleus accumbens, prefrontal cortex, and caudate nucleus and locomotor activity are modulated in dose–response characteristics by methylphenidate. Brain Research. 2006a Feb 16;1073:164-74.
29. Yang PB, Swann AC, Dafny N. Chronic administration of methylphenidate produces neurophysiological and behavioral sensitization. Brain Research. 2007a May 11;1145:66-80.
30. Dafny N, Yang PB. The role of age, genotype, sex, and route of acute and chronic administration of methylphenidate: a review of its locomotor effects. Brain Research Bulletin. 2006 Feb 15;68(6):393-405.
31. Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Research Reviews. 1993 Sep 1;18(3):247-91.
32. Everitt BJ, Robbins TW. From the ventral to the dorsal striatum: devolving views of their roles in drug addiction. Neuroscience & Biobehavioral Reviews. 2013 Nov 1;37(9):1946-54.
33. Gardner EL. Brain reward mechanisms. Substance Abuse: A comprehensive Textbook. 1997:51-85.
34. Kalivas PW. Introduction: The circuitry mediating the translation of motivational stimuli into adaptive motor responses. Limbic Motor Circuits and Neuropsychiatry. 1993: pp.237-288.
35. Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. Journal of Neuroscience. 2006 Jun 14;26(24):6583-8.
36. Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Progress in Neurobiology. 1998 Apr 24;54(6):679-720.
37. Woolverton WL, Johnson KM. Neurobiology of cocaine abuse. Trends in pharmacological sciences. 1992 Jan 1;13:193-200.
38. Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences. 1988 Jul 1;85(14):5274-8.
39. Ikemoto S. Dopamine reward circuitry: two projection systems from the ventral midbrain to the nucleus accumbens–olfactory tubercle complex. Brain Research Reviews. 2007 Nov 1;56(1):27-78.
40. Haber SN, Knutson B. The reward circuit: linking primate anatomy and human imaging. Neuropsychopharmacology. 2010 Jan;35(1):4-26.
41. Haber SN. Chapter 1—anatomy and connectivity of the reward circuit. H of R and DM. Academic Press, New York. 2017; pp:3-19.
42. Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988 Nov 4;242(4879):715-23.
43. Oldehinkel M, Beckmann CF, Pruim RH, Van Oort ES, Franke B, Hartman CA, et al. Attention-deficit/ hyperactivity disorder symptoms coincide with altered striatal connectivity. Biological psychiatry: cognitive neuroscience and neuroimaging. 2016 Jul 1;1(4):353-63.
44. Podet A, Lee MJ, Swann AC, Dafny N. Nucleus accumbens lesions modulate the effects of methylphenidate. Brain Research Bulletin. 2010 Jul 30;82(5-6):293-301.
45. Wanchoo SJ. Role of Dopamine of Nucleus Accumbens in Behavioral Sensitization to Methylphenidate. UT Graduate School of Biomedical Sciences at Houston: Dissertations and Theses. 2010 May 1:14.
46. Bell K, Kalivas PW. Context-specific cross-sensitization between systemic cocaine and intra-accumbens AMPA infusion in the rat. Psychopharmacology. 1996 Jun;127(1):377-83.
47. Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue- induced cocaine craving. American Journal of Psychiatry. 1999 Jan 1;156(1):11-8.
48. Claussen CM, Chong SL, Dafny N. Selective bilateral lesion to caudate nucleus modulates the acute and chronic methylphenidate effects. Pharmacology Biochemistry and Behavior. 2012 Apr 1;101(2):208-16.
49. Hemby SE, Horman B, Tang W. Differential regulation of ionotropic glutamate receptor subunits following cocaine self-administration. Brain research. 2005 Dec 7;1064(1-2):75-82.
50. Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral tegmental area glutamate neurons: electrophysiological properties and projections. Journal of Neuroscience. 2012 Oct 24;32(43):15076-85.
51. Jones Z, Dafny N. Acute and chronic dose–response effect of methylphenidate on ventral tegmental area neurons correlated with animal behavior. Journal of Neural Transmission. 2014 Mar 1;121(3):327-45.
52. Kalivas PW, Duffy P. Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. Journal of Neurochemistry. 1998 Apr;70(4):1497- 502.
53. King N, Floren S, Kharas N, Thomas M, Dafny N. Glutaminergic signaling in the caudate nucleus is required for behavioral sensitization to methylphenidate. Pharmacology Biochemistry and Behavior. 2019 Sep 1;184:172737.
54. Pierce RC, Bell K, Duffy P, Kalivas PW. Repeated cocaine augments excitatory amino acid transmission in the nucleus accumbens only in rats having developed behavioral sensitization. Journal of Neuroscience. 1996 Feb 15;16(4):1550-60.
55. Pierce RC, Reeder DC, Hicks J, Morgan ZR, Kalivas PW. Ibotenic acid lesions of the dorsal prefrontal cortex disrupt the expression of behavioral sensitization to cocaine. Neuroscience. 1998 Feb 12;82(4):1103-14.
56. Pierce RC, Kalivas PW. A circuitry model of the expression of behavioral sensitization to amphetaminelike psychostimulants. Brain Research Reviews. 1997 Oct 1;25(2):192-216.
57. Reid MS, Berger SP. Evidence for sensitization of cocaine-induced nucleus accumbens glutamate release. Neuroreport. 1996 May 1;7(7):1325-9.
58. Wanchoo SJ, Swann AC, Dafny N. Descending glutamatergic pathways of PFC are involved in acute and chronic action of methylphenidate. Brain Research. 2009 Nov 16;1301:68-79.
59. Wanchoo SJ, Lee MJ, Swann AC, Dafny N. Bilateral six-hydroxydopamine administration to PFC prevents the expression of behavioral sensitization to methylphenidate. Brain Research. 2010 Feb 2;1312:89-100.
60. White SR, Harris GC, Imel KM, Wheaton MJ. Inhibitory effects of dopamine and methylenedioxymethamphetamine (MDMA) on glutamate-evoked firing of nucleus accumbens and caudate/putamen cells are enchanced following cocaine self-administration. Brain Research. 1995 May 29;681(1-2):167-76.
61. Claussen CM, Dafny N. Caudate neuronal recording in freely behaving animals following acute and chronic dose response methylphenidate exposure. Pharmacology Biochemistry and Behavior. 2015 Sep 1;136:21-30.
62. Gaytan O, al-Rahim S, Swann A, Dafny N. Sensitization to locomotor effects of methylphenidate in the rat. Life sciences. 1997 Jul 18;61(8):PL101-7.
63. Karim TJ, Reyes-Vazquez C, Dafny N. Comparison of the VTA and LC response to methylphenidate: A concomitant behavioral and neuronal study of adolescent male rats. Journal of Neurophysiology. 2017 Sep 1;118(3):1501-14.
64. Karim TJ, Aksel C, Kharas N, Reyes-Vasquez C, Dafny N. Caudate nucleus neurons participate in methylphenidate function: Behavioral and neuronal recordings from freely behaving adolescent rats. Brain Rsearch Bulletin. 2018 Sep 1;142:241-52.
65. Yang PB, Swann AC, Dafny N. Chronic pretreatment with methylphenidate induces cross-sensitization with amphetamine. Life sciences. 2003b Oct 17;73(22):2899- 911.
66. Yang PB, Swann AC, Dafny N. Chronic methylphenidate modulates locomotor activity and sensory evoked responses in the VTA and NAc of freely behaving rats. Neuropharmacology. 2006b Sep 1;51(3):546-56.
67. Yang PB, Swann AC, Dafny N. Methylphenidate treated at the test cage—dose-dependent sensitization or tolerance depend on the behavioral assay used. Critical Reviews™ in Neurobiology. 2007b;19(2):59-77.
68. Yang PB, Cuellar III DO, Swann AC, Dafny N. Age and genetic strain differences in response to chronic methylphenidate administration. Behavioural Brain Research. 2011 Mar 17;218(1):206-17.
69. Paxinos G, Watson C. The rat brain in stereotaxic coordinates: hard cover edition. Elsevier; 2006 Nov 2.
70. Kouvelas ED, Mitsacos A, Angelatou F, Hatziefthimiou A, Tsiotos P, Voukelatou G. Glutamate receptors in mammalian cerebellum: Alterations in human ataxic disorders and cerebellar mutant mice. InCerebellar degenerations: Clinical Neurobiology 1992 (pp. 123-137). Springer, Boston, MA.
71. Meldrum BS. Glutamate as a neurotransmitter in the brain: review of physiology and pathology. The Journal of Nutrition. 2000 Apr 1;130(4):1007S-15S.
72. McEntee WJ, Crook TH. Glutamate: its role in learning, memory, and the aging brain. Psychopharmacology. 1993 Jul;111(4):391-401.
73. Shigeri Y, Seal RP, Shimamoto K. Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Research Reviews. 2004 Jul 1;45(3):250- 65.
74. Schwarcz R, Hökfelt T, Fuxe K, Jonsson G, Goldstein M, Terenius L. Ibotenic acid-induced neuronal degeneration: a morphological and neurochemical study. Experimental Brain Research. 1979 Oct;37(2):199-216.
75. Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P. Methylphenidate-induced dendritic spine formation and ?FosB expression in nucleus accumbens. Proceedings of the National Academy of Sciences. 2009 Feb 24;106(8):2915-20.
76. McGinty VB, Grace AA. Timing-dependent regulation of evoked spiking in nucleus accumbens neurons by integration of limbic and prefrontal cortical inputs. Journal of neurophysiology. 2009 Apr;101(4):1823-35.
77. Perez MF, Ford KA, Goussakov I, Stutzmann GE, Hu XT. Repeated cocaine exposure decreases dopamine D2-like receptor modulation of Ca2+ homeostasis in rat nucleus accumbens neurons. Synapse. 2011 Feb;65(2):168- 80.
78. Salgado S, Kaplitt MG. The nucleus accumbens: a comprehensive review. Stereotactic and Functional Neurosurgery. 2015;93(2):75-93.
79. Zaborszky L, Alheid GF, Beinfeld MC, Eiden LE, Heimer L, Palkovits M. Cholecystokinin innervation of the ventral striatum: a morphological and radioimmunological study. Neuroscience. 1985 Feb 1;14(2):427-53.
80. Zahm DS, Brog JS. On the significance of subterritories in the “accumbens” part of the rat ventral striatum. Neuroscience. 1992 Oct 1;50(4):751-67.
81. Brog JS, Salyapongse A, Deutch AY, Zahm DS. The patterns of afferent innervation of the core and shell in the “accumbens” part of the rat ventral striatum: immunohistochemical detection of retrogradely transported fluoro-gold. Journal of Comparative Neurology. 1993 Dec 8;338(2):255-78.
82. Groenewegen HJ, Wright CI, Beijer AV, Voorn P. Convergence and segregation of ventral striatal inputs and outputs. Annals of the New York Academy of Sciences. 1999 Jun;877(1):49-63.
83. Phillipson OT, Griffiths AC. The topographic order of inputs to nucleus accumbens in the rat. Neuroscience. 1985 Oct 1;16(2):275-96.
84. Williams DJ, Crossman AR, Slater P. The efferent projections of the nucleus accumbens in the rat. Brain Research. 1977 Jul 15;130(2):217-27.
85. Anden NE, Jackson DM. Locomotor activity stimulation in rats produced by dopamine in the nucleus accumbens: potentiation by caffeine. Journal of Pharmacy and Pharmacology. 1975 Sep;27(9):666-70.
86. Austin MC, Kalivas PW. Dopaminergic involvement in locomotion elicited from the ventral pallidum/substantia innominata. Brain Research. 1991 Feb 22;542(1):123-31.
87. Boye SM, Grant RJ, Clarke PB. Disruption of dopaminergic neurotransmission in nucleus accumbens core inhibits the locomotor stimulant effects of nicotine and D-amphetamine in rats. Neuropharmacology. 2001 May 1;40(6):792-805.
88. Cadoni C, Solinas M, Di Chiara G. Psychostimulant sensitization: differential changes in accumbal shell and core dopamine. European Journal of Pharmacology. 2000 Jan 24;388(1):69-76.
89. Cornish JL, Kalivas PW. Repeated cocaine administration into the rat ventral tegmental area produces behavioral sensitization to a systemic cocaine challenge. Behavioural Brain Research. 2001 Nov 29;126(1-2):205-9.
90. Heidbreder C, Feldon J. Amphetamine-induced neurochemical and locomotor responses are expressed differentially across the anteroposterior axis of the core and shell subterritories of the nucleus accumbens. Synapse. 1998 Aug;29(4):310-22.
91. Hooks MS, Jones GH, Liem BJ, Justice Jr JB. Sensitization and individual differences to IP amphetamine, cocaine, or caffeine following repeated intracranial amphetamine infusions. Pharmacology Biochemistry and Behavior. 1992 Nov 1;43(3):815-23.
92. Kalivas PW, Weber BR. Amphetamine injection into the ventral mesencephalon sensitizes rats to peripheral amphetamine and cocaine. Journal of Pharmacology and Experimental Therapeutics. 1988 Jun 1;245(3):1095-102.
93. Paulson PE, Robinson TE. Sensitization to systemic amphetamine produces an enhanced locomotor response to a subsequent intra-accumbens amphetamine challenge in rats. Psychopharmacology. 1991 May;104(1):140-1.
94. Perugini M, Vezina P. Amphetamine administered to the ventral tegmental area sensitizes rats to the locomotor effects of nucleus accumbens amphetamine. Journal of Pharmacology and Experimental Therapeutics. 1994 Aug 1;270(2):690-6.
95. Swanson CJ, Heath S, Stratford TR, Kelley AE. Differential behavioral responses to dopaminergic stimulation of nucleus accumbens subregions in the rat. Pharmacology Biochemistry and Behavior. 1997 Dec 1;58(4):933-45.
96. Vezina P, Stewart J. Amphetamine administered to the ventral tegmental area but not to the nucleus accumbens sensitizes rats to systemic morphine: lack of conditioned effects. Brain Research. 1990 May 14;516(1):99-106.
97. Joyce EM, Stinus L, Iversen SD. Effect of injections of 6-OHDA into either nucleus accumbens septi or frontal cortex on spontaneous and drug-induced activity. Neuropharmacology. 1983 Sep 1;22(9):1141-5.
98. Kelly PH, Iversen SD. Selective 60HDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. European Journal of Pharmacology. 1976 Nov 1;40(1):45- 56
99. Kelsey JE, Willmore EJ. Electrolytic lesions of the nucleus accumbens enhance locomotor sensitization to nicotine in rats. Behavioral neuroscience. 2006 Jun;120(3):600.
100. Koob GF, Stinus L, Le Moal M. Hyperactivity and hypoactivity produced by lesions to the mesolimbic dopamine system. Behavioural Brain Research. 1981 Nov 1;3(3):341-59.
101. Sellings LH, Clarke PB. 6-Hydroxydopamine lesions of nucleus accumbens core abolish amphetamine-induced conditioned activity. Synapse. 2006 May;59(6):374-7.
102. Todtenkopf MS, Carreiras T, Melloni Jr RH, Stellar JR. The dorsomedial shell of the nucleus accumbens facilitates cocaine-induced locomotor activity during the induction of behavioral sensitization. Behavioural Brain Research. 2002 Apr 11;131(1-2):9-16.
103. Weiner I, Gal G, Rawlins JN, Feldon J. Differential involvement of the shell and core subterritories of the nucleus accumbens in latent inhibition and amphetamineinduced activity. Behavioural Brain Research. 1996 Nov 1;81(1-2):123-33.
104. Garcia-Keller C, Martinez SA, Esparza MA, Bollati F, Kalivas PW, Cancela LM. Cross-sensitization between cocaine and acute restraint stress is associated with sensitized dopamine but not glutamate release in the nucleus accumbens. European Journal of Neuroscience. 2013 Mar;37(6):982-95.
105. Shin JH, Adrover MF, Wess J, Alvarez VA. Muscarinic regulation of dopamine and glutamate transmission in the nucleus accumbens. Proceedings of the National Academy of Sciences. 2015 Jun 30;112(26):8124-9.
106. Sellings LH, Clarke PB. Segregation of amphetamine reward and locomotor stimulation between nucleus accumbens medial shell and core. Journal of Neuroscience. 2003 Jul 16;23(15):6295-303.
107. Zahm DS. Functional-anatomical implications of the nucleus accumbens core and shell subterritories. Annals of the New York Academy of Sciences. 1999 Jun;877(1):113-28.
108. Baracz SJ, Rourke PI, Pardey MC, Hunt GE, McGregor IS, Cornish JL. Oxytocin directly administered into the nucleus accumbens core or subthalamic nucleus attenuates methamphetamine-induced conditioned place preference. Behavioural Brain Research. 2012 Mar 1;228(1):185-93.
109. Jang JK, Kim WY, Cho BR, Lee JW, Kim JH. Microinjection of ghrelin in the nucleus accumbens core enhances locomotor activity induced by cocaine. Behavioural Brain Research. 2013 Jul 1;248:7-11.
110. Hu A, Lai M, Wei J, Wang L, Mao H, Zhou W, et al. The effect of electroacupuncture on extinction responding of heroin-seeking behavior and FosB expression in the nucleus accumbens core. Neuroscience Letters. 2013 Feb 8;534:252-7.
111. Waselus M, Flagel SB, Jedynak JP, Akil H, Robinson TE, Watson Jr SJ. Long-term effects of cocaine experience on neuroplasticity in the nucleus accumbens core of addiction-prone rats. Neuroscience. 2013 Sep 17;248:571- 84.
112. Feja M, Hayn L, Koch M. Nucleus accumbens core and shell inactivation differentially affects impulsive behaviours in rats. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2014 Oct 3;54:31-42.
113. West EA, Carelli RM. Nucleus accumbens core and shell differentially encode reward-associated cues after reinforcer devaluation. Journal of Neuroscience. 2016 Jan 27;36(4):1128-39.
114. Yee J, Famous KR, Hopkins TJ, McMullen MC, Pierce RC, Schmidt HD. Muscarinic acetylcholine receptors in the nucleus accumbens core and shell contribute to cocaine priming-induced reinstatement of drug seeking. European Journal of Pharmacology. 2011 Jan 15;650(2-3):596-604.