Abstract
We present a case of apical hypertrophic cardiomyopathy (AHCM), also known as Yamaguchi syndrome, in a young female with no family history. She presented after recurrent syncopal events with cyanosis, palpitations and was found to have diffuse T-wave inversions and a pattern of left ventricular hypertrophy on ECG. Echocardiogram and cardiac magnetic resonance imaging (MRI) showed evidence of hypertrophic cardiomyopathy (HCM) localized to the apical wall segments along with late gadolinium enhancement (LGE), without left ventricular outflow tract (LVOT) obstruction. The patient required telemetry to monitor for malignant arrythmias. She required electrophysiology evaluation and genetic testing, which was significant for an autosomal dominant mutation localized to filamin C. The patient received a tilt table test and was positive for cardioinhibitory and vasodepressor response but did not have evidence of malignant arrhythmia. She received an implantable loop recorder (ILR) for long term monitoring and was stable for discharge with close out-patient follow up. The patient requires re-imaging with cardiac MRI due to risk of possible early progression of fibrosis and sudden cardiac death (SCD) with the identified genetic mutation. She was not able to tolerate beta blockers for medical therapy and is being monitored off medication. AHCM is a rare form of HCM characterized by left ventricular apical wall thickness measured to be greater than 15mm and may be identified by an “ace of spades” pattern of the left ventricular cavity during systole on echocardiogram and cardiac MRI. If associated with LGE, this can be a risk factor for SCD. It is also autosomal dominant in inheritance and can be associated with sarcomere protein mutations. The treatment of AHCM is similar to HCM, focusing on preserving left ventricular preload by slowing heart rate and contractility with beta blockers, calcium channel blockers, disopyramide. Patients that are refractory to medical therapy may benefit from surgical myomectomy or alcohol septal ablation. The long-term prognosis of AHCM has been studied to be favorable compared to other forms of HCM. This unique case offers insight into the diagnosis and treatment strategies of AHCM and offers a case to reappraise the potential challenges in identifying this condition without an established family history.
Keywords
Cardiac biomarkers, Cardiomyopathies, Cardiovascular imaging, Pediatric cardiology
Introduction
Hypertrophic Cardiomyopathy (HCM) is a common disorder characterized by asymmetric hypertrophy of the septal myocardium. HCM is present in 1 out of 500 people of the general adult population and it equally affects both men and women [1]. A subset of HCM includes hypertrophic obstructive cardiomyopathy (HOCM), which is characterized by myocyte disarray that contributes to a left ventricular outflow tract (LVOT) dynamic obstruction, most commonly due to systolic anterior motion of the anterior leaflet of the mitral valve. HOCM is prevalent in the young population and is the most common cause of sudden cardiac death in well-trained athletes [2]. The pathophysiology behind HOCM deals with the asymmetric septal hypertrophy contributing to a transient LVOT obstruction, obstructing blood flow and leading to impaired ventricular filling with resultant decreased left ventricular cavity size. This contributes to reduced cardiac output and can contribute to coronary ischemia, leading to septal micro-infarctions, contributing to development of chronic scar tissue, and introducing substrates for reentrant circuits, contributing to the initiation of malignant ventricular arrhythmias. HOCM is commonly a genetic disorder, which is autosomal dominant in inheritance, consisting of mutations in genes that code for sarcomere proteins including β-myosin heavy chain, actin, titin, troponin, and tropomyosin [3]. The condition is characterized by variable expressivity/penetrance, so it has a wide range of clinical presentations. Despite the prevalence of HOCM, most patients are not diagnosed until a significant cardiac event has occurred, being that a plurality of patients may be asymptomatic or have nonspecific symptoms that can obfuscate the diagnosis. When there is an underlying dynamic obstructive process, patients can experience symptoms such as presyncope, dizziness, syncope, angina, palpitations (as a result of arrhythmias), and dyspnea. Symptoms are often worsened with exertion. Despite the wide presence of HOCM, about 60% of familial cases have the identified sarcomere protein mutations, whereas the remaining one third of patients have unidentified genetic mutations that are still being investigated [4]. Filamin C has been observed in previous studies to be a novel mutation involved in the pathogenesis of HOCM, due to impaired function of the filamin protein localized to the sarcomere which facilitates the interaction between the sarcomere proteins myotilin and myozenin. Mutations in Filamin C have also been associated with myofibrillar myopathy and are hypothesized to be a risk factor for sudden cardiac death (SCD) [5].
Identification on examination may be difficult as HOCM is often a silent disease. The characteristic murmur is a systolic crescendo-decrescendo, which is often loudest at the third intercostal space at the left lower sternal border. The identification of the characteristic murmur can be accentuated with provocative maneuvers that will intensify the obstruction. In conditions that decrease both preload and afterload, such as with a Valsalva maneuver, being dehydrated, or rising suddenly from sitting position, there will be less volume present in the left ventricular heart chamber, despite an increased oxygen demand due to wall strain. This contributes to a decreased cardiac output and coronary ischemia, which may precipitate dangerous arrhythmias. In conditions that increase both preload and afterload, the LVOT obstruction is lessened, and symptoms may improve. Maneuvers, such as handgrip, Valsalva release, squatting, and leg-raising while supine will diminish the intensity of the murmur by increasing the preload and/or afterload, therefore improving diastolic filling of the left ventricular cavity—thereby improving the degree of obstruction and improving symptoms [6]. In severe long-term untreated cases, adults with HOCM may have early signs of heart failure as initial symptoms after several decades, as HOCM can be silent for a very long time, whereas pediatric patients are at an elevated risk of heart failure, more likely to develop within the first few years after diagnosis [7,8]. Signs and symptoms of heart failure that may be seen include but are not limited to—dyspnea on exertion, orthopnea, paroxysmal nocturnal dyspnea, jugular venous distension, peripheral pitting edema, hepatosplenomegaly, ascites [9]. Despite these findings, one of the most common initial presetting symptoms of HOCM is when the patient presents in a malignant ventricular rhythm, such as ventricular tachycardia or ventricular fibrillation.
Although the hypertrophy in HCM is often localized to the interventricular septum, there are identified subtypes of HCM that can have atypical presentations, even in the absence of an established family history. Apical hypertrophic cardiomyopathy (AHCM), also known as Yamaguchi Syndrome, is a rare form of HCM in which hypertrophy is localized to the apical walls [10]. It can be associated with the same clinical features— syncope, angina, dyspnea, palpitations, cyanosis, exercise intolerance, and in severe cases early heart failure symptoms, malignant arrhythmias, and sudden cardiac death. On diagnostic testing, ECG is characterized by diffuse T-wave inversions and can present with larger QRS voltages that are higher than those defined by typical voltage criteria for left ventricular hypertrophy (LVH), although this is not always present. While AHCM typically has tall R waves in the left precordial leads (V4-V5), studies have shown that the maximal R wave amplitude in lead V5 in AHCM can be significantly higher than 30 mm [11]. There can also be early R wave progression if the hypertrophy is more localized to the apex and does not significantly involve the ventricular septum, although these ECG patterns in AHCM poorly correlate with its anatomical features and can vary between patients [12]. On echocardiogram, there is increased thickness of the left ventricular wall apical regional, which is measured to be greater than 15 mm [13]. There may also be associated findings of a ventricular aneurysm, which would increase the risk of thromboembolic events. Echocardiogram with contrast administration can also demonstrate the classic “ace of spades” pattern of the left ventricular cavity during systole when in presence of AHCM, due to obliteration of the apex. Although echocardiography is used to assess for LVOT obstruction and is one of the initial studies to diagnose AHCM, cardiac magnetic resonance imaging (MRI) is a more sensitive modality for diagnosis, as it provides high resolution images that span the entire left ventricle, including the apex. On MRI, the same left ventricular apical wall thickness greater than 15 mm, along with an apical to basal wall thickness ratio >1.5 are both suggestive of AHCM [14]. MRI also has the additional benefit of incorporating late gadolinium enhancement (LGE) in the study, which if positive is suggestive of fibrosis, and may serve as a substrate for arrhythmogenic foci. LGE is a risk factor for SCD in these patients. Pharmacological treatment for patients with AHCM is similar to that for HOCM, which deals with preload and heart rate optimization for patients. Beta blockers are beneficial in that they can help diminish the noradrenergic effects on the heart and decrease both the heart rate and contractility. This will increase the diastolic filling time and allow more blood to enter and remain in the left ventricle during diastole, thereby increasing preload and augmenting cardiac output [15]. In patients unable to tolerate beta blockers, calcium channel blockers can achieve the same effect through a different mechanism: by preventing calcium influx, they are able to help suppress the sinus and atrioventricular nodes, which in turn contributes to a decreased heart rate and contractility. These effects improve diastolic filling time and thus augment cardiac output. Disopyramide has also been utilized for its negative chronotropic effects if the patient cannot tolerate either beta blockers or calcium channel blockers [16].
Some patients are not able to tolerate medical therapy due to possible unwanted medication side effects, which include but are not limited to—hypotension, fatigue, dizziness, nausea, bronchospasm, sexual dysfunction, and pedal edema; the latter symptom although to a lesser degree with non-dihydropyridine calcium blockers in contrast to dihydropyridine calcium channel blockers.
In patients that do not improve with medical therapy, surgical therapy may be the next viable option. Surgical myectomy has been performed for more than five decades and has been associated with very good long-term outcomes in HOCM; by excising cardiac tissue around the hypertrophied segment, the procedure is able to alleviate the LVOT obstruction and alleviate the symptoms of HOCM [17]. The mitral insufficiency that many patients with HOCM experience almost always resolves following surgical myectomy. Despite its benefits, myectomy is an open-heart surgery and may not be indicated in elderly patients with significant comorbidities. Damage to the electrical conduction system may occur, and a small percentage of patients have required permanent pacemaker implantation as a result. A nonsurgical option is an alcohol septal ablation, which is a transcatheter procedure whereby injection of alcohol into the asymmetric hypertrophied segment of the heart facilitates the infarction and shrinkage of the pathogenic hypertrophied tissue. The atrophy and scarring of the interventricular septum alleviate the obstructive physiology of HOCM, resulting in lower LVOT gradients. This, being a less invasive procedure, has a quicker recovery time when compared to surgical myectomy, and has similar long term clinical outcomes. However, complete alleviation of obstruction is less likely with alcohol septal ablation, and patients may require re-intervention. Despite these measured differences, it should also be noted that surgeons may introduce significant selection bias by choosing only optimal candidates for septal reduction surgery, by analyzing clinical criteria, hemodynamic parameters, medical comorbidities, and anatomic criteria as judged by the surgeon. The patients that are not deemed ideal surgical candidates are instead likely to be referred for less invasive alcohol septal ablation. This selection practice inadvertently introduces bias, as the surgical cohort will likely be younger, while the higher risk cohort is preferentially directed towards ablation therapy [7]. Lastly, when patients are identified as being high risk for malignant arrhythmia, they require risk stratification to determine whether they should receive implantable cardioverter-defibrillator (ICD) placement for sudden cardiac death prevention.
Case Description
A previously healthy 17-year-old Hispanic female with a past medical history of obesity, menorrhagia, generalized anxiety disorder (GAD), and major depressive disorder (MDD) initially presented to the outpatient clinic with new-onset syncope in October 2023. The first syncopal episode occurred while brushing her teeth in June 2023, and she felt the sensation that despite this, her lips were dry and also felt palpitations and then suffered a loss of consciousness without injuries to her face.
The family repositioned the patient in a chair, and she briefly regained consciousness. She recalls a “heart racing” sensation that was present immediately prior to fainting, which was still present; then, the patient lost consciousness for a second time. She was unconscious for less than one minute each time; however, the entire period of presyncope/syncope lasted about 10-15 minutes. During each episode of syncope, witnesses reported that the patient appeared pale with perioral cyanosis. She completely recovered; therefore, the parents did not seek medical attention until after the 3rd syncopal episode, which happened five days prior to her presentation at the cardiology office. Her 3rd episode lasted approximately 5 minutes. Shortly prior to this episode, she was hanging balloons outside for a birthday party, when she again felt the prodromal symptoms of dry lips, palpitations and then suddenly lost consciousness. She fell to the ground slowly and her family was concerned that she hit her head, although she had no evidence of trauma or any injuries. She denied ever feeling chest pain associated with the syncopal episodes and she endorsed that she had felt palpitations in the past that were separate from her syncopal episodes and are occasionally accompanied by a shortness of breath. On her visits with her primary medical provider for the last decade, it was communicated that she never had a history of syncope in the past prior to these episodes. Her family history was completely negative for premature death, sudden cardiac death (SCD), dysrhythmia, and congenital heart disease. The only significant family history is hypertension and diabetes in her mother, and von Willebrand Disease in her sister. On the physical exam, lungs were clear to auscultation bilaterally, and cardiac exam revealed regular rate, rhythm, normal S1 with a physiologic splitting of S2, and there was no presence of murmurs or gallops on auscultation. An ECG done in the office, Figure 1 demonstrated diffuse T-wave inversions and high voltage R-waves in the precordial leads, meeting criteria for left ventricular hypertrophy.
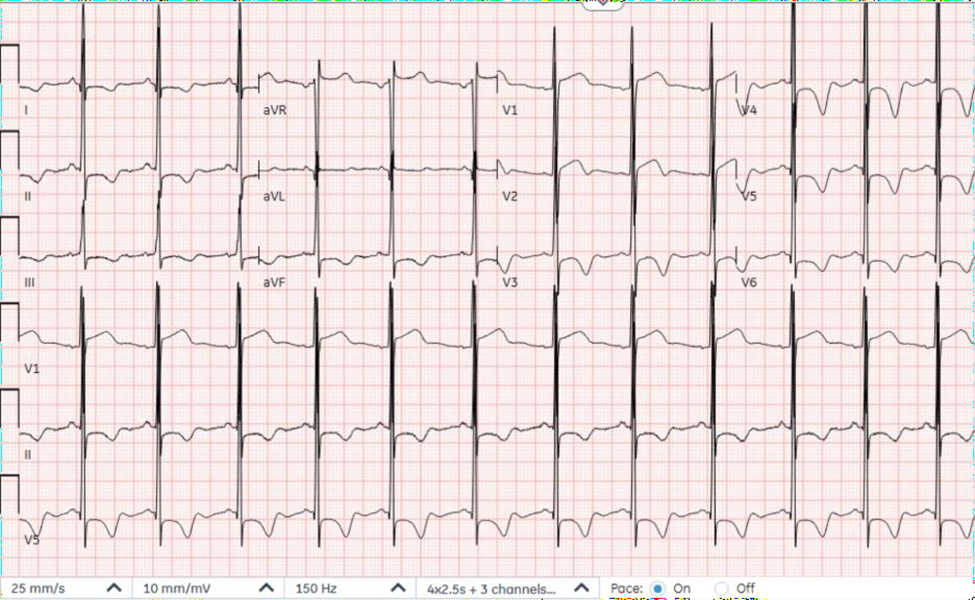
Figure 1. Electrocardiogram demonstrating characteristic diffuse large T-wave inversions, seen best in anterolateral precordial leads, along with a biphasic T-wave in Lead V2 and evidence of left ventricular hypertrophy by voltage criteria.
The patient was referred to pediatric cardiology for further evaluation of these ECG findings. The echocardiogram performed in the cardiology office the following day showed evidence of hypertrophic cardiomyopathy, primarily affecting the apical wall segments. A hyperdynamic left ventricular systolic function noted. There was no evidence of systolic anterior motion of the mitral valve, nor LVOT obstruction. Based upon the above, a diagnosis of hypertrophic cardiomyopathy (HCM) was made (Figures 2–4). The cardiologist was concerned that ventricular arrhythmia could have resulted caused her syncopal episodes. Therefore, exercise restriction and admission to the pediatric intensive care unit was recommended for further evaluation of HCM via telemetry monitoring, genetic testing, cardiac MRI to evaluate for fibrosis, and electrophysiology (EP) consult for possible ICD device placement for secondary prevention.
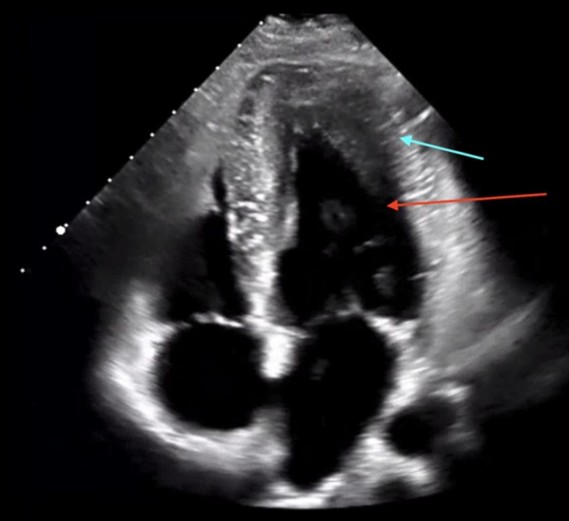
Figure 2. Transthoracic echocardiogram, Apical 4 Chamber view showing the localized several apical segment hypertrophy of the left ventricle (blue arrow). There is also the characteristic “ace of spades” geometry, reflective of systolic obliteration of the apex (red arrow).
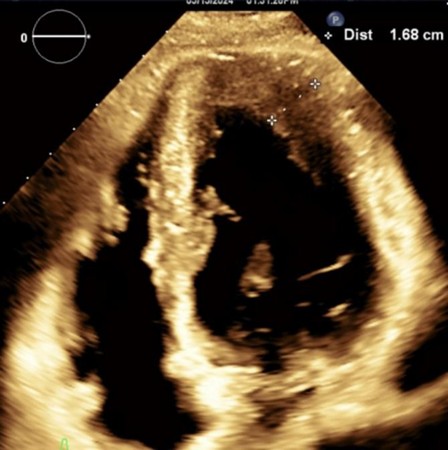
Figure 3. Transthoracic Echocardiogram, 2 chamber parasternal short axis view, demonstrating apical hypertrophy. The left ventricular wall measured 1.68 cm, greater than 15 mm, coinciding with the diagnosis of hypertrophic cardiomyopathy.
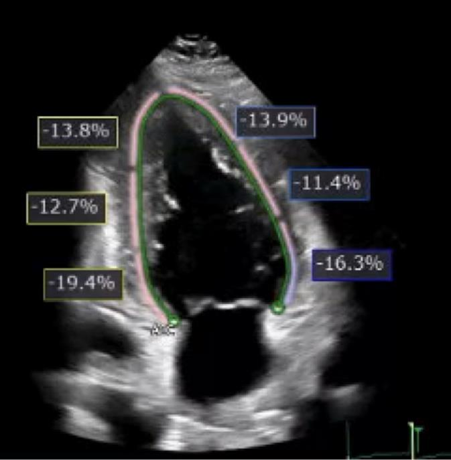
Figure 4. Transthoracic echocardiogram with evidence of reduced global longitudinal strain of -14% with reduced strain in all apical and mid-cavitary wall segments.
A cardiac MRI with contrast was consistent with the apical variety of hypertrophic cardiomyopathy (Figure 5). The maximum left ventricular wall thickness at the apex was 2.2 cm without evidence of left ventricular aneurysm or wall motion abnormalities. There was also a mild amount of LGE in the mid myocardium in the distal infero-septal/infero-apical regions, suggestive of interstitial fibrosis. A tilt table test to evaluate for vasovagal syncope was performed by cardiac EP, which returned positive. The test reproduced syncope with similar prodrome to prior episodes. As a result, the patient was started on 25 mg metoprolol succinate once daily. ICD placement was deferred in the absence of demonstrable ventricular arrhythmias, and the presence of a cardioinhibitory etiology of her vasovagal syncope. An internal loop recorder (ILR) was implanted for long-term monitoring. Post- discharge, the patient was unable to tolerate metoprolol due to burning sensation and skin discomfort and was switched to nadolol. Unfortunately, this patient had a similar reaction to nadolol in addition to itching of her eyes, and beta blockers as a class were discontinued. EP cardiology determined that she can be monitored off beta blockers at present. Genetic testing detected an autosomal dominant variant of FLNC (filamin C, an actin-binding protein primarily expressed in heart and skeletal muscle) considered to be of unknown clinical significance, for which the patient is heterozygous. Per EP cardiology recommendations, a cardiac MRI will be repeated to assess for apical aneurysm and increased LGE from the prior study, as there are reports of this variant being associated with a higher incidence of SCD [18].
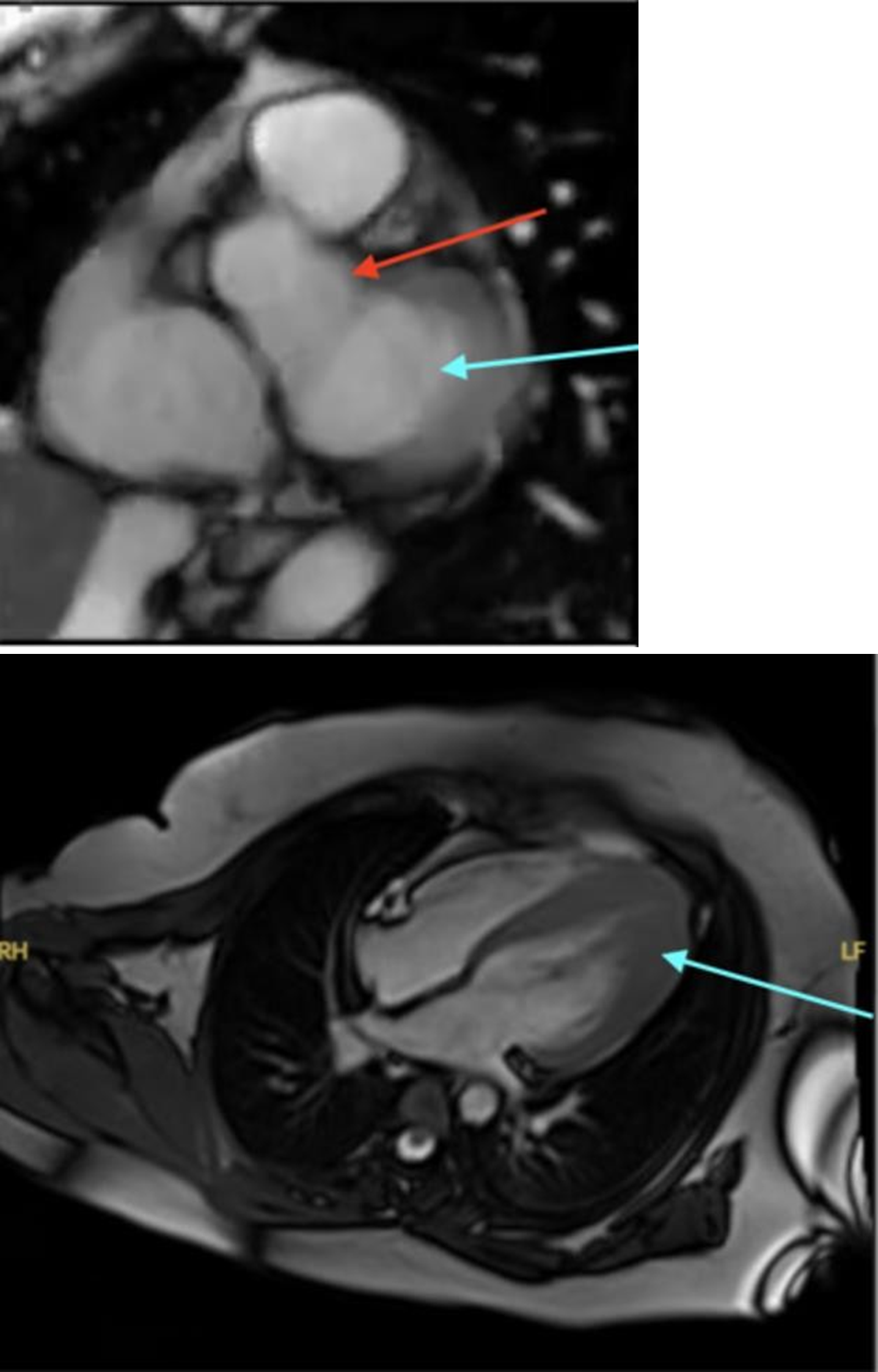
Figure 5. Cardiac MRI. The first image demonstrates a normal cardiac MRI, with no evidence of left ventricular hypertrophy. The blue arrow demarcates the left ventricle and the red arrow identifies the ascending aorta. The normal cardiac MRI listed was not from our patient of the case discussion, rather it was obtained from the Cardiac MRI learning center [19]. The second image was a cardiac MRI of the patient from the case discussion, demonstrating the presence of apical hypertrophic cardiomyopathy as outlined by the blue arrow.
Discussion
Yamaguchi Syndrome, also known as Apical Hypertrophic Cardiomyopathy (AHCM), is an atypical form of HCM that consists of an asymmetrical hypertrophy of the left ventricle localized to the apical wall segments. It has similar manifestations of symptoms compared to other forms of HCM, and similar physical exam findings. AHCM is also a disease with genetic origins, with the most commonly implicated being the autosomal dominant sarcomere protein genes—MYBPC3 and MYH7 [20]. Despite there being a strong genetic predisposition for this disease, only 13% of patients of AHCM have been identified to be genotype positive, raising concern and importance toward encouraging more genetic testing to further evaluate for familial predisposition for this disease. Other studies have further examined that up to 40% of HCM patients have been identified to fall into a nonfamilial form of the disease and have more benign courses compared to the mutation-positive varieties [21].
The diagnosis of Yamaguchi Syndrome involves certain unique findings, including—diffuse, large T-wave inversions on ECG, greater than 1 millivolt in any lead, along with concurrent echocardiography or cardiac MRI findings of apical wall thickness measured to be greater than 15 mm, and a ratio of apical to basal/posterior wall thickness to be greater than 1.5. There can also be the characteristic “ace of spades” pattern of the left ventricle on ventriculography or cardiac imaging, indicative of obliteration of the left ventricular apex [22]. As a result of the localization of the hypertrophy to the apical wall segments, there can be less of an occurrence of a dynamic LVOT obstruction.
The findings of LGE on imaging are a marker of tissue fibrosis because of the hypertrophic remodeling in the ventricle, which without appropriate treatment may continue to worsen with time, leading to worsening heart function. This has been identified to be a marker of clinical severity and disease progression in these patients [23]. Other markers of clinical severity and progression in this disease, may reflect evidence of left ventricular aneurysm because of turbulent and static blood flow. There may also be a complete end systolic ventricular cavity obliteration, as a result of worsening apical hypertrophy leading to complete collapse of the apical cavity during ventricular systole. AHCM has been studied to have similar complications to other forms of HOCM, with the risk of worsening heart failure, progression of coronary ischemia, infarction, stroke, arrhythmias, and sudden cardiac death. There have been a multitude of large Asian cohort studies that have consistently shown that AHCM has a favorable prognosis compared to other subtypes of HCM [24,25]. However, analysis of AHCM in North American patients has been limited and underpowered in previous studies, therefore there remains a need for further investigation in this area and careful longitudinal follow up in these patients [26].
Lastly, the presence of Filamin C (FLNC) gene mutation in this patient is problematic and associated with increased risk of SCD. FLNC encodes an actin binding protein that is crucial for maintaining sarcomere integrity during contraction. When this gene is mutated, there is decreased ability for the protein to bind to actin, therefore leading abnormal protein folding, aggregation and hypertrophy, thereby disrupting sarcomere architecture [5]. These protein aggregates that are localized in the apical region will predispose to apical hypertrophy and myocyte dysfunction, contributing to long term fibrosis, which is associated with arrhythmogenic substrate formation and a heightened risk of SCD [27].
Conclusion
Yamaguchi syndrome, or apical hypertrophic cardiomyopathy (AHCM), is a unique form of hypertrophic cardiomyopathy characterized by asymmetric apical wall thickening rather than the more commonly-affected septal region. This unique presentation makes it more challenging to diagnose, as many patients are asymptomatic, and the patients who do have symptoms can exhibit a broad range of findings. Their symptoms span from mild fatigue to severe chest pain and syncope (reflecting malignant arrhythmias or myocardial ischemia), further complicating the clinical vignette. The diagnosis of this disease requires imaging with echocardiography or MRI to identify the localized apical wall thickness in proportion to other areas of the ventricular myocardium. The management of Yamaguchi syndrome remains difficult due to the heterogeneous nature of the disease, with treatment strategies focusing on symptom control and prevention of disease progression with various medications. By optimizing diastolic filling time with beta blockers, calcium channel blockers, or disopyramide, the left ventricular outflow tract obstruction is mitigated. Patients that are refractory to medical management may benefit from invasive procedures like septal myectomy and alcohol septal ablation. The unique structural features of this disease require careful monitoring and individualized care plans for optimal treatment. Genetic testing plays a crucial role in both the diagnosis and management of Yamaguchi syndrome, not only identifying the unique mutations in the sarcomere proteins but also allowing for family screening and personalized treatment strategies. As genetic research continues to evolve, analyzing and understanding the underlying genetic mutations in Yamaguchi syndrome will become increasingly important for early diagnosis, risk stratification, and the development of targeted therapies. Ultimately, the complexity of Yamaguchi syndrome emphasizes the need for ongoing research, heightened clinical awareness, and the integration of genetic testing to improve patient outcomes.
References
2. Malhotra A, Sharma S. Hypertrophic Cardiomyopathy in Athletes. Eur Cardiol. 2017 Dec;12(2):80–2.
3. Marian AJ. Molecular Genetic Basis of Hypertrophic Cardiomyopathy. Circ Res. 2021 May 14;128(10):1533–53.
4. Watkins H, Ashrafian H, Redwood C. Inherited cardiomyopathies. N Engl J Med. 2011 Apr 28;364(17):1643–56.
5. Valdés-Mas R, Gutiérrez-Fernández A, Gómez J, Coto E, Astudillo A, Puente DA, et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat Commun. 2014 Oct 29;5:5326.
6. Houston BA, Stevens GR. Hypertrophic cardiomyopathy: a review. Clin Med Insights Cardiol. 2015 Jan 26;8(Suppl 1):53–65.
7. Writing Committee Members; Ommen SR, Ho CY, Asif IM, Balaji S, Burke MA, Day SM, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR Guideline for the Management of Hypertrophic Cardiomyopathy: A Report of the American Heart Association/American College of Cardiology Joint Committee on Clinical Practice Guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324–405.
8. Marston NA, Han L, Olivotto I, Day SM, Ashley EA, Michels M, et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur Heart J. 2021 May 21;42(20):1988–96.
9. Harris C, Croce B, Munkholm-Larsen S. Hypertrophic obstructive cardiomyopathy. Ann Cardiothorac Surg. 2017 Jul;6(4):429.
10. Giri A, Acharya S, Kamat S, Shukla S, Kumar S. Yamaguchi Syndrome: A Hidden Masquerader of Ischemic Heart Disease. Cureus. 2022 Jun 29;14(6):e26439.
11. Tao Y, Xu J, Bako SY, Yao X, Yang D. Usefulness of ECG to differentiate apical hypertrophic cardiomyopathy from non-ST elevation acute coronary syndrome. BMC Cardiovasc Disord. 2020 Jun 23;20(1):306.
12. Hughes RK, Thornton GD, Malcolmson JW, Pierce I, Khoury S, Hornell A, et al. Accurate diagnosis of apical hypertrophic cardiomyopathy using explainable advanced electrocardiogram analysis. Europace. 2024 Mar 30;26(4):euae093.
13. Paluszkiewicz J, Krasinska B, Milting H, Gummert J, Pyda M. Apical hypertrophic cardiomyopathy: diagnosis, medical and surgical treatment. Kardiochir Torakochirurgia Pol. 2018 Dec;15(4):246–53.
14. Hughes RK, Knott KD, Malcolmson J, Augusto JB, Mohiddin SA, Kellman P, et al. Apical Hypertrophic Cardiomyopathy: The Variant Less Known. J Am Heart Assoc. 2020 Mar 3;9(5):e015294.
15. Östman-Smith I. Beta-Blockers in Pediatric Hypertrophic Cardiomyopathies. Rev Recent Clin Trials. 2014;9(2):82–5.
16. Adler A, Fourey D, Weissler-Snir A, Hindieh W, Chan RH, Gollob MH, et al. Safety of Outpatient Initiation of Disopyramide for Obstructive Hypertrophic Cardiomyopathy Patients. J Am Heart Assoc. 2017 May 26;6(6):e005152.
17. Nishimura RA, Seggewiss H, Schaff HV. Hypertrophic Obstructive Cardiomyopathy: Surgical Myectomy and Septal Ablation. Circ Res. 2017 Sep 15;121(7):771–83.
18. Filamin C Registry Consortium; Gigli M, Stolfo D, Barbati G, Graw S, Chen SN, Merlo M, et al. Arrhythmic Risk Stratification of Carriers of Filamin C Truncating Variants. JAMA Cardiol. 2025 Apr 1;10(4):359–69.
19. Cardiac MRI. “Home ‑ Cardiac MRI.” CardiacMRI.com, 2025 (Accessed 22 Sept. 2025). Available at: https://cardiacmri.com/.
20. Gruner C, Care M, Siminovitch K, Moravsky G, Wigle ED, Woo A, et al. Sarcomere protein gene mutations in patients with apical hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2011 Jun;4(3):288–95.
21. Ingles J, Burns C, Bagnall RD, Lam L, Yeates L, Sarina T, et al. Nonfamilial Hypertrophic Cardiomyopathy: Prevalence, Natural History, and Clinical Implications. Circ Cardiovasc Genet. 2017 Apr;10(2):e001620.
22. Ono R, Iwahana T, Kobayashi Y. Ace-of-spades with tear drop sign in apical hypertrophic cardiomyopathy. QJM. 2021 Apr 27;114(2):130–1.
23. Yin Y, Hu W, Zhang L, Wu D, Yang C, Ye X. Clinical, echocardiographic and cardiac MRI predictors of outcomes in patients with apical hypertrophic cardiomyopathy. Int J Cardiovasc Imaging. 2022 Mar;38(3):643–51.
24. Li J, Fang J, Liu Y, Wei X. Apical hypertrophic cardiomyopathy: pathophysiology, diagnosis and management. Clin Res Cardiol. 2024 May;113(5):680–93.
25. An S, Fan C, Yan L, Cai C, Yang Y, Zhai S, et al. Comparison of Long-Term Outcome between Apical and Asymmetric Septal Hypertrophic Cardiomyopathy. Cardiology. 2017;136(2):108–14.
26. Eriksson MJ, Sonnenberg B, Woo A, Rakowski P, Parker TG, Wigle ED, et al. Long-term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002 Feb 20;39(4):638–45.
27. Celeghin R, Cipriani A, Bariani R, Bueno Marinas M, Cason M, Bevilacqua M, et al. Filamin-C variant-associated cardiomyopathy: A pooled analysis of individual patient data to evaluate the clinical profile and risk of sudden cardiac death. Heart Rhythm. 2022 Feb;19(2):235–43.