Keywords
Intravascular lymphoma, Rituximab, Circulating tumor DNA, Hemophagocytosis, Random skin biopsy
Introduction
Intravascular B cell lymphoma (IVBCL) is notoriously difficult to diagnose as the clinical manifestations are protean, and the patterns seen with routine labs and imaging are non-specific [1]. Furthermore, the disease follows an aggressive course and is often fatal within a matter of weeks to months from symptom onset, unless recognized and treated appropriately [2]. This has historically meant that diagnosis was made at autopsy for many patients. Over the past few decades, however, scientific and clinical literature have slowly accumulated to better characterize and raise clinical awareness of this disease. In this paper, we will review the characteristics that make this diagnosis challenging, and then discuss new and emerging diagnostic avenues.
Biology & Classification of IVBCL
IVBCL is an aggressive extranodal large B-cell lymphoma, characterized by the presence of malignant lymphocytes exclusively in the lumen of small and occasionally medium sized vessels [3]. While the pathophysiology of this disease is not fully understood, the fulminant clinical course (despite relatively low-volume disease) is likely a result of microvascular ischemia in the affected organs due to diffuse microvascular occlusion and dysfunction caused by malignant lymphocytes [4]. This distinctive intravascular tropism seems to derive both from the presence of molecules that facilitate endothelial adhesion, and also the absence of adequate levels of chemokine receptors and metalloproteinases involved in extravasation and parenchymal invasion [5].
It is a rare disease, with an incidence estimated at less than 1 in 10 million in the United States [4]. Previously, the disease was categorized into two variants, Western and Asian, based on different disease characteristics observed in case series from these regions [3,7-9]. The Western phenotype was associated with cutaneous lesions, more prominent neurologic symptoms and lower rates of organomegaly and marrow involvement [3,7]. This category also included a cutaneous variant, typically seen in younger woman and associated with better clinical outcomes. The Asian variant, described in case series mostly from Japan, is characterized by hepatosplenomegaly, thrombocytopenia, frequent bone marrow involvement and sometimes hemophagocytosis [8] (Table 1).
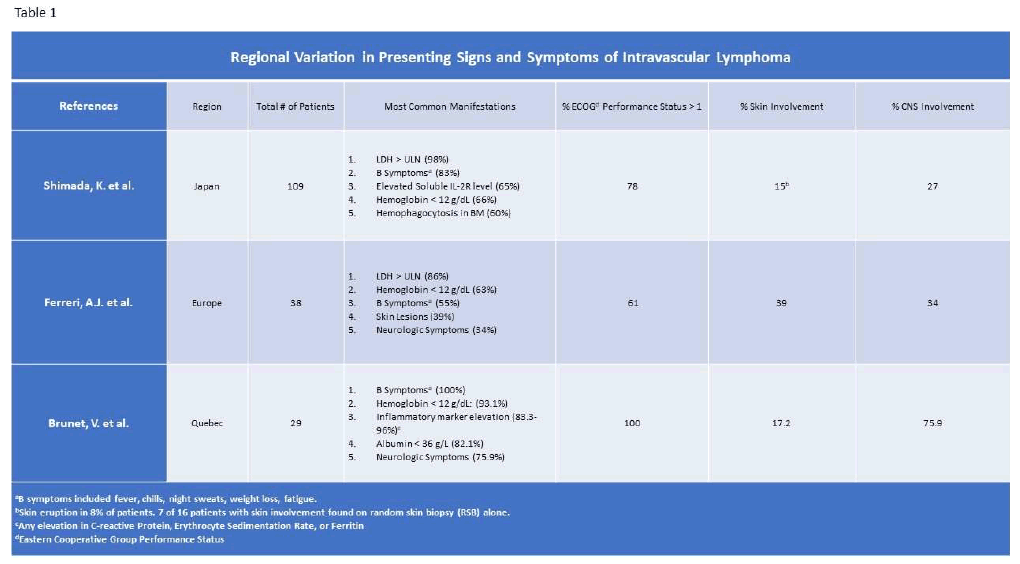
However, the utility of these diagnostic categories has been lessened by the facts that: [1] there is significant clinical overlap between the two groups, and [2] the classification arose from differences between case series and not distinctions based on individual patient characteristics within those series. Therefore, the most recent WHO classification uses the clinical phenotypes of classical, cutaneous, and hemophagocytosis-associated, as opposed to regional/ethnic distinctions [10].
Within the classical and hemophagocytic variants, constitutional symptoms including fever and rapid clinical decline (progressive organ dysfunction, weakness, mental status changes, etc.) are common and seen in more than 60% of patients [3,7,8,10]. Organ-specific symptoms vary widely depending on sites of disease involvement. The most frequently reported neurologic manifestations are encephalopathy, stroke-like syndromes and seizures, but lesions mapping to the spinal cord and even peripheral nerves are also seen [7,11-15]. Involvement of the vasculature of lungs, GI tract and endocrine organs leads to their respective organ failure syndromes [4,7]. A variety of different cutaneous lesions are reported in leads to their respective organ failure syndromes [4,7]. Western series. The most frequent findings are plaques and macules, but cutaneous lesions can have wideranging presentations, including nodules, telangiectasias and peau d’orange. [7,16]. The hemophagocytic variant presents with symptoms consistent with hemophagocytic lymphohistiocytosis, including fever, hepatosplenomegaly and cytopenias [8].
Treatment
The treatment for IVBCL is typically combination chemoimmunotherapy including an anthracycline and rituximab [17-21]. The most common combination chemoimmunotherapy described in the medical literature is R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone). Two of the largest published retrospective series of patients treated with rituximabcontaining regimens from Japan and Europe showed a 2-year overall survival (OS) of 66% and 3-year OS of 81%, respectively [20,21]. Median follow up time in these cohorts were 17 and 23 months, respectively, in those patients treated with rituximab-containing regimens. Conversely, the 3-year OS reported in the North American case series was only 42.7% in those that received similar therapy at a median follow up of 10.8 months [11]. Much of the variance in these survival estimates likely stems from difference in study methodology (i.e., how patients were identified and selected for inclusion). However, as with the overwhelming majority of B-cell lymphomas, the addition of rituximab does appear to have improved outcomes compared to historical controls [18,20,21].
Evaluation for CNS disease at diagnosis, including magnetic resonance imaging (MRI) of the brain and CSF analysis, is recommended given the high rates of CNS involvement [3]. However, MRI findings can be absent or non-specific, and CSF abnormalities are similarly variable [11]. Thus, given the high rates of CNS relapse, CNS prophylaxis with intrathecal chemotherapy or high dose methotrexate has been routinely recommended regardless of initial CNS diagnostic evaluation [20,21].
Given the high rates of both systemic and CNS relapse, some clinicians have pursued high-dose therapy with autologous stem cell transplant (HDT-ASCT) as either an upfront strategy in first remission, or at relapse [22]. Given the paucity and retrospective nature of the data for this approach, it is not possible to make any strong recommendations as to the benefit and risk tradeoffs for ASCT. However. HDT-ASCT is at least feasible in a subset of patients and has been associated with long-term remission. To our knowledge, there are no publications on the use of novel immune therapies such as checkpoint inhibitors and Chimeric Antigen Receptor T Cells (CAR-T) in patients with IVBCL. In an evaluation of IVBCL cells from the NIH archives, a subset was found to express PDL1 [23].
Although treatment outcomes appear to have improved over time, some of this may be accounted for by earlier diagnosis as well as improvements in supportive care. Perhaps the greatest challenge to treatment is the diagnostic delay, such that many patients have a poor performance status (Eastern Cooperative Oncology Group performance status >1 in 80-100% [7,8,11] and significant organ dysfunction at the time of presentation and diagnosis. Thus, rapid accurate diagnosis is the crucial and rate-limiting step in the management of IVBCL [2].
A Formidable Diagnostic Challenge
Whereas diagnosis of cutaneous IVBCL may be made relatively early with biopsy of the affected skin, a variety of factors contribute to the difficulty of diagnosing classical and hemophagocytic IVBCL. One significant contributor is the non-specific clinical presentation. In the majority (65%+) of non-cutaneous variant cases, patients present with an inflammatory milieu of fever and constitutional symptoms, accompanied by a variety of organ-specific symptoms as detailed above [7,9]. These myriad symptoms overlap with a variety of other syndromes, including infections (particularly endocarditis, zoonoses, fungal and mycobacterial), rheumatologic syndromes such as vasculitis or connective tissue disease, and other neoplastic diseases and paraneoplastic neurologic syndromes. There are rarely particular signs or symptoms that can quickly and reliably distinguish between these conditions, and thus patients with IVBCL frequently undergo an extensive workup.
Laboratory testing in this disease also frequently yields indeterminate results. A high lactate dehydrogenase (LDH) level appears to be fairly sensitive for the disease, but is non-specific [24]. Anemia is the most common hematologic abnormality [7], although thrombocytopenia is seen with the hemophagocytic variant [8]. Even a finding such as a myeloperoxidase-antineutrophilic cytoplasmic autoantibody (MPO+ ANCA), which has a reported specificity of up to 98% in the diagnosis of vasculitis, cannot necessarily exclude the presence of an underlying IVBCL [13,25].
Given the lack of tumor aggregate, cross-sectional imaging is non-specific and can demonstrate organomegaly, airspace opacities and pulmonary nodules, or a variety of patterns on CNS imaging [11]. There is some evidence to support the utility of 18F-FDG PET, which can show patchy and diffusely increased uptake in the lungs, bone and reticuloendothelial organs [26,27]. While these patterns are non-specific, they can be suggestive. PET imaging can also help rule out discrete masses suggestive of an alternative diagnosis. While there is no prospective validation of Lugano criteria for staging or evaluating treatment response in IVBCL specifically, decreased FDG uptake has been seen following treatment with chemotherapy [26]. A simplified approach to endof- treatment response is reasonable given the lack of validated Lugano criteria in this setting, and was used by Shimada et al. in their retrospective analysis. This approach classified post-treatment response as complete response, progressive disease, or stable disease, and utilized information obtained from 18F-FDG PET (with or without concurrent CT). Complete response in this setting is resolution of clinical symptoms, as well as imaging and lab abnormalities. Progressive disease is either new disease symptoms/lesions and/or progression in known lesions or symptoms. Stable disease is categorized as neither meeting criteria for progressive disease nor complete response in this study [20]. Thus, obtaining 18F-FDG PET (with or without concurrent CT) may be helpful with initial diagnosis, and is prudent once diagnosis is confirmed as a possible method of tracking response to therapy.
As a result of the protean presentations of IVBCL, a frequent scenario with this disease is a patient presenting with constitutional symptoms and a rapidly declining clinical course, but without pathognomonic findings to suggest the underlying diagnosis. Initial testing will be consistent with a non-specific inflammatory process. Depending on the capabilities of a given medical facility, many of the diagnostic labs as part of the initial workup (i.e., fungal and atypical bacterial studies, extended rheumatologic antibody testing, paraneoplastic antibody panels from the CNS) may require days to weeks to be resulted.
Biopsy
In this scenario, a diagnostic biopsy takes on particular importance for the diagnosis of IVBCL and exclusion of other malignant, inflammatory, or infectious etiologies. Even if IVBCL is suspected, there are challenges to identifying a biopsy site with acceptable safety and high diagnostic yield. In IVBCL, peripheral blood flow cytometry historically has had had a sensitivity less than 10% [1], despite the fact that the disease is exclusively present in/ around blood vessels. Bone marrow involvement ranges from 30-75% percent, with higher percentages reported in Japanese cohorts [9]. Thus, while these two testing modalities will likely be undertaken early in the course of the workup, negative testing does not rule out the presence of IVBCL.
IVBCL can theoretically be detected with the biopsy of any affected organ with small blood vessels involvement [9,16,28-30]. However, given the low tumor volume, false negative biopsies are common, and the pathologist should be notified about the suspicion for IVBCL to look for subtle lymphocyte aggregates in the lumen of small vessels. The malignant cells appear similar to other high-grade large B cell lymphomas, with prominent nucleoli, scant cytoplasm, and frequent mitotic figures. The immunophenotypic expression of malignant lymphocytes commonly includes CD79a, CD19 and CD20, as well as Mum1 and Bcl-2 [31]. Recurrent cytogenetic abnormalities in chromosomes 1, 6q and 18 have been described in the largest karyotypic analysis, which included a total of 29 patients [32]. Recurrent mutations in MYD88 and CD79A have been described, and are further detailed in the Liquid Biopsy section.
An additional practical challenge is that biopsy of many potential sites (reticuloendothelial organs, lungs, endocrine organs, kidney, brain) is intrinsically risky, and particularly in patients who may already be medically unstable or with pre-existing co-morbidities. The average age of patients with IVBCL is around 60-70 years, where preexisting medical issues are common and increase the risk of both complications and rapid clinical decline. Biopsy is further complicated by factors such as thrombocytopenia, respiratory failure and encephalopathy. Coagulopathy or frank disseminated intravascular coagulation (DIC) can occur during the clinical course of IVBCL with a documented frequency as high as 25% of patients in one of the larger cohorts studied [33,34]. Encephalopathy can be a particularly challenging disease manifestation, as it complicates the process of informed consent and may necessitate the use of sedation or intubation for diagnostic procedures, both of which can worsen the encephalopathy. A final consideration is that the diagnostic ambiguity with these cases often makes the risk/benefit calculation of a biopsy more difficult to estimate.
Given all these difficulties with securing a diagnosis of IVBCL in a deteriorating patient, we are in dire need of alternative diagnostic avenues. Two particular techniques may be of use in this regard: random biopsies of unaffected skin, and so-called “liquid biopsies” that interrogate cell free (and ideally) circulating tumor DNA.
Random Skin Biopsy
There are case reports of IVBCL diagnosis obtained via biopsy of a visible skin lesion [28,35]. However, more recent literature from Japan has claimed that random skin biopsy (RSB) of apparently uninvolved skin can be a relatively low-risk diagnostic tool. Matsue et al [36], reviewed 114 cases in which RSB was performed to evaluate for IVBCL. Thirty-three patients were eventually diagnosed with IVBCL, 26 having a diagnosis established with a single incisional biopsy, deep enough to include subcutaneous fat. They calculated a sensitivity of 78% with a specificity near 99% for IVBCL. That same group published a research letter advocating specifically for deep incisional biopsies, based on retrospective review of their pathology specimens which demonstrated that 46% of specimens had IVBCL present only in the vasculature of subcutaneous fat, with no apparent involvement of the dermis [37]. Another report notes that a random biopsy of normal appearing, non-FDG avid skin was still diagnostic for IVBCL [27].
There are some caveats to these data, however. This series came from a single hospital system in Japan. It is uncertain whether this approach would be as useful with a different set of clinicians, with varying degrees of expertise and training in performing deep incisional skin biopsy, or with a different patient demographic. 29% of patients in the cohort were eventually diagnosed with IVBCL, suggesting that the clinical team may have been astutely identifying patients with a higher-pretest probability of IVBCL. Additionally, biopsy sensitivity for IVBCL at other sites, such as bone marrow, has varied significantly between studies performed in Western (~30%), Japanese (~65%) and non-Japanese East Asian (~38%) cohorts [9]. In the Matsue study, 65% of the patients diagnosed with IVBCL by skin biopsy also had detectable disease in the bone marrow, and 80% with negative skin biopsies were found to have marrow disease. Sensitivity of RSB may similarly vary between different patient populations, and there are no other comparable large case series of RSB, particularly outside of Japan. The limited data available from studies in the United States [38] and Quebec [11] are insufficient to judge the sensitivity or specificity of RSB for the diagnosis of IVBCL in these respective populations. However, given the relatively low risk of RSB compared to other biopsy sites, this may be a reasonable diagnostic approach, particularly if a non-diagnostic bone marrow biopsy has already been performed.
Liquid Biopsy: cfDNA/ctDNA Assessment
The development of next generation sequencing (NGS) has spurred advances in the management of both hematologic and solid organ malignancies [39-41]. Sequencing can identify prognostic genetic variations and abnormalities, and may be predictive of response to targeted therapies (e.g., FLT3 inhibitors in acute myeloid leukemia (AML) or epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer). Cell-free DNA (cfDNA) is nonencapsulated DNA released by damaged or dying cells (e.g., during apoptosis) that can be detected in the peripheral blood, with higher concentrations found in patients with a variety of medical conditions including autoimmune diseases, infection and cancers [42]. Furthermore, in many cancer patients, a subset of cfDNA derived from the tumor, known as circulating tumor DNA (ctDNA), can be detected. Assessment of ctDNA obtained from the peripheral blood of cancer patients can allow for rapid genetic assessment of prognostic and predictive genetic variations. A theoretical advantage of this approach is that the sample can be obtained with a peripheral blood draw, avoiding some of the delays inherent in obtaining and processing a solid biopsy specimen. Contrarily, the relative novelty of ctDNA analysis does lead to outstanding questions about optimal preanalytical and analytic approaches.
In solid tumor oncology, ctDNA is on the road to becoming a standard of care evaluation in locally advanced colorectal cancer following resection, in order to better delineate which patients are most likely to develop recurrent disease. In one study of those patients with resected stage II or III colorectal cancer, patients with detectable ctDNA displayed a significantly shorter 2-year relapsefree survival, time to recurrence and overall survival than ctDNA-negative patients. Eleven (92%) of ctDNA-positive patients developed recurrence compared to 9 (7%) of ctDNA-negative patients [43]. ctDNA assessment in patients with DLBCL has demonstrated some prognostic value with regards to response and risk of relapse [44,45]. As these techniques come into broader use and we deepen our understanding of the genetic underpinnings of hematologic malignancies, ctDNA assessment could be useful for a variety of hematologic malignancies [46,47]. Given the difficulties in securing a diagnosis of IVBCL with solid tissue biopsy, the use of cf/ctDNA assessment is an intriguing possibility.
The utility of cf/ctDNA assessment in the diagnosis of IVBCL was explored in a recent research letter [47]. In this study, advanced sequencing of 8 genes was performed on both tumor-derived DNA (tdDNA) and serum and plasma cfDNA from 9 patients with IVBCL. These genes, including MYD88 and CD79B, had previously been shown to harbor recurrent mutations in IVBCL [48]. In samples from all 9 patients, a detectable mutation in at least one of the 8 genes was detected via cfDNA analysis of the serum and/or plasma. cfDNA was found to be more sensitive at detecting these mutations than tdDNA, and higher variant allele frequencies (VAF) were seen in cfDNA samples as well. Longitudinal sampling demonstrated that cfDNA VAF of presumed pathogenic MYD88 and CD79B mutations correlated with disease activity, such that sicker patients with higher clinical disease burden had higher VAFs. Taken together, this study provides preliminary evidence that the cfDNA detected was ctDNA, and that similar analysis could become a useful diagnostic tool for IVBCL.
There are numerous hurdles blocking the routine availability and use of cfDNA/ctDNA technology for the diagnosis of IVBCL. More extensive studies of IVBCL are needed to define the range of genetic abnormalities and establish genetic fingerprints that are sufficiently sensitive and specific to be clinically useful, as they are unlikely to be independently diagnostic. As noted above, MYD88 has been identified as a potential variant in IVBCL subsets. However, it is better known for its association with Waldenstrom’s Macroglobulinemia, highlighting the inherent specificity problems in this rare disease. In the near future, it is unlikely that circulating tumor DNA assays optimized and validated for IVBCL will be available, given the rarity of the disease. Additionally, many medical facilities may not have access to clinical labs able to perform these studies in a timeframe that would be useful for treatment. However, as the usage of cfDNA/ctDNA technology increases for more common lymphomas (particularly diffuse large B-cell lymphoma), the knowledge from these data sets may be extrapolated to IVBCL. Thus, as liquid biopsy techniques advance, these technologies could become a useful adjunctive tool to aid in the early recognition and treatment of IVBCL.
Conclusion
Intravascular B cell lymphoma remains a formidable opponent: difficult to diagnose, with aggressive but nebulous disease manifestations. Early recognition and evaluation for IVBCL is likely a significant factor in improving prognosis with this disease. A comprehensive and astute work-up must balance a rapid and intensive diagnostic evaluation against the risks for iatrogenic harm. Some of the newer techniques of cfCNA and ctDNA may provide opportunities to expand the sensitivity and specificity of testing for IVBCL with minimal direct patient risks. Logistics and availability of these advanced genetic techniques will continue to be barriers until the framework of ctDNA is better established. R-CHOP remains the standard of care for IVBCL, although further advances in treatment will have the opportunity to parallel those of aggressive B-cell non-Hodgkin lymphomas. Some case series continue to support relatively high percentages of 2- and 3-year overall survivals in patients able to be appropriately diagnosed and treated, further highlighting the importance of rapid diagnosis and initiation of therapy for this rare and aggressive disease.
References
2. Fonkem E, Lok E, Robison D, Gautam S, Wong ET. The natural history of intravascular lymphomatosis. Cancer Med. 2014; 3 (4): 1010-24.
3. Ponzoni M, Ferreri AJ, Campo E, Facchetti F, Mazzucchelli L, Yoshino T, et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: proposals and perspectives from an international consensus meeting. Journal of Clinical Oncology. 2007 Jul 20;25(21):3168-73.
4. Fukunaga H, Kawashima K, Kumakawa H, Hashimoto Y, Takahashi Y. An autopsy of intravascular large B-cell lymphoma with hemophagocytic syndrome. JRSM open. 2017 Apr;8(5):2054270417695054.
5. Ponzoni M, Campo E, Nakamura S. Intravascular large B-cell lymphoma: a chameleon with multiple faces and many masks. Blood, The Journal of the American Society of Hematology. 2018 Oct 11;132(15):1561-7.
6. Rajyaguru DJ, Bhaskar C, Borgert AJ, Smith A, Parsons B. Intravascular large B-cell lymphoma in the United States (US): a population-based study using Surveillance, Epidemiology, and End Results program and National Cancer Database. Leukemia & Lymphoma. 2017 Sep 2;58(9):1-9.
7. Ferreri AJ, Campo E, Seymour JF, Willemze R, Ilariucci F, Ambrosetti A, et al. Intravascular lymphoma: clinical presentation, natural history, management and prognostic factors in a series of 38 cases, with special emphasis on the ‘cutaneous variant’1. British Journal of Haematology. 2004 Oct;127(2):173-83.
8. Murase T, Yamaguchi M, Suzuki R, Okamoto M, Sato Y, Tamaru JI, et al. Intravascular large B-cell lymphoma (IVLBCL): a clinicopathologic study of 96 cases with special reference to the immunophenotypic heterogeneity of CD5. Blood. 2007 Jan 15;109(2):478-85.
9. Ferreri AJ, Dognini GP, Campo E, Willemze R, Seymour JF, Bairey O, et al. Variations in clinical presentation, frequency of hemophagocytosis and clinical behavior of intravascular lymphoma diagnosed in different geographical regions. Haematologica. 2007 Apr 1;92(4):486-92.
10. Nakamura S. Intravascular large B-cell lymphoma. WHO classification of tumors of haematopoietic and lymphoid tissues. C.E. Swerdlow SH, Harris NL, et al, Editor. 2017, IARC: Lyon, France. p. 317-318.
11. Brunet V, Marouan S, Routy JP, Hashem MA, Bernier V, Simard R, et al. Retrospective study of intravascular large B-cell lymphoma cases diagnosed in Quebec: a retrospective study of 29 case reports. Medicine. 2017 Feb;96(5): e5985.
12. Maiese A, La Russa R, De Matteis A, Frati P, Fineschi V. Post-mortem diagnosis of intravascular large B-cell lymphoma. Journal of International Medical Research. 2020 Jun;48(6):0300060520924262.
13. Usuda D, Arahata M, Temaru R, Iinuma Y, Kanda T, Hayashi S. Autopsy-proven intravascular lymphoma presenting as rapidly recurrent strokes. Case Reports in Oncology. 2016;9(1):148-53.
14. Sugiyama A, Kobayashi M, Daizo A, Suzuki M, Kawashima H, Kagami SI, et al. Diffuse cerebral vasoconstriction in an intravascular lymphoma patient with a high serum MPO-ANCA level. (1349-7235 (Electronic)).
15. Fonkem E, Dayawansa S, Stroberg E, Lok E, Bricker PC, Kirmani B, et al. Neurological presentations of intravascular lymphoma (IVL): meta-analysis of 654 patients. BMC Neurology. 2016 Dec 1;16(1):9.
16. Röglin J, Böer A. Skin manifestations of intravascular lymphoma mimic inflammatory diseases of the skin. British Journal of Dermatology. 2007 Jul;157(1):16-25.
17. Ferreri AJ, Campo E, Ambrosetti A, Ilariucci F, Seymour JF, Willemze R, et al. Anthracycline-based chemotherapy as primary treatment for intravascular lymphoma. Annals of Oncology. 2004 Aug 1;15(8):1215- 21.
18. Ferreri AJ, Dognini GP, Bairey O, Szomor A, Montalbán C, Horvath B, et al. The addition of rituximab to anthracycline-based chemotherapy significantly improves outcome in ‘Western’patients with intravascular large B-cell lymphoma. British Journal of Haematology. 2008 Oct;143(2):253-7.
19. Shimada K, Murase T, Matsue K, Okamoto M, Ichikawa N, Tsukamoto N, et al. Central nervous system involvement in intravascular large B-cell lymphoma: A retrospective analysis of 109 patients. Cancer Science. 2010 Jun;101(6):1480-6.
20. Shimada K, Matsue K, Yamamoto K, Murase T, Ichikawa N, Okamoto M, et al. Retrospective analysis of intravascular large B-cell lymphoma treated with rituximab-containing chemotherapy as reported by the IVL study group in Japan. Journal of Clinical Oncology. 2008 Jul 1;26(19):3189-95.
21. Ferreri AJ, Dognini GP, Govi S, Crocchiolo R, Bouzani M, Bollinger CR, et al. Can rituximab change the usually dismal prognosis of patients with intravascular large B-cell lymphoma? . Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008 Nov 1;26(31):5134-5136.
22. Meissner J, Finel H, Dietrich S, Boumendil A, Kanfer E, Laboure G, et al. Autologous hematopoietic stem cell transplantation for intravascular large B-cell lymphoma: the European Society for Blood and Marrow Transplantation experience. Bone Marrow Transplantation. 2017 Apr;52(4):650-2.
23. Gupta GK, Jaffe ES, Pittaluga S. A study of PDL1 expression in intravascular large B cell lymphoma: correlation with clinical and pathological features. Histopathology. 2019 Aug;75(2):282-6.
24. Masaki Y, Dong L, Nakajima A, Iwao H, Miki M, Kurose N, et al. Intravascular large B cell lymphoma: proposed of the strategy for early diagno sis and treatment of patients with rapid deteriorating condition. (1865-3774 (Electronic)).
25. Naffaa ME, Rozin AP, Horowitz N, Ben-Itzhak O, Braun-Moscovici Y, et al. Diffuse large B cell lymphoma mimicking granulomatosis with polyangiitis. Case Reports in Rheumatology. 2016 May 2016: p. 1041787-1041787.
26. Shi X, Huo L, Sun J, Ding J, Xing H, Wang T, et al. The Role of 18F-FDG PET/CT in the Assessment of Patients with Intravascular Large B-Cell Lymphoma. Journal of Nuclear Medicine. 2019 May 1;60(supplement 1):1249.
27. Miura Y, Tsudo M. Fluorodeoxyglucose-PET/CT for diagnosis of intravascular large B-cell lymphoma. InMayo Clinic Proceedings 2010 Aug 1 (Vol. 85, No. 8, pp. e56-e57).
28. Gill S, Melosky B, Haley L, ChanYan C. Use of random skin biopsy to diagnose intravascular lymphoma presenting as fever of unknown origin. The American Journal of Medicine. 2003 Jan 1;114(1):56-8.
29. Raza M, Qayyum S, Raza S, Goorha S. Intravascular B-cell lymphoma: an elusive diagnosis. Journal of Clinical Oncology. 2012 May 20;30(15): e144-5.
30. D’Angelo CR, Ku K, Gulliver J, Chang J. Intravascular large B-cell lymphoma presenting with altered mental status: A case report. World Journal of Clinical Oncology. 2019 Dec 24;10(12):402-8.
31. Orwat DE, Batalis NI. Intravascular large B-cell lymphoma. Archives of Pathology & Laboratory Medicine. 2012 Mar;136(3):333-8.
32. Klairmont MM, Cheng J, Martin MG, Gradowski JF. Recurrent cytogenetic abnormalities in intravascular large B-cell lymphoma. American Journal of Clinical Pathology. 2018 May 31;150(1):18-26.
33. Lui PC, Wong GK, Poon WS, Tse GM. Intravascular lymphomatosis. Journal of Clinical Pathology. 2003 Jun 1;56(6):468-70.
34. Shimada K, Kinoshita T, Naoe T, Nakamura S. Presentation and management of intravascular large B-cell lymphoma. The lancet oncology. 2009 Sep 1;10(9):895- 902.
35. Yunoki M, Suzuki K, Uneda A, Yoshino K. A case of intravascular lymphoma presenting as myelopathy diagnosed with a skin biopsy. Surgical Neurology International. 2015;6(Suppl 13): S367.
36. Matsue K, Abe Y, Kitadate A, Miura D, Narita K, Kobayashi H, et al. Sensitivity and specificity of incisional random skin biopsy for diagnosis of intravascular large B-cell lymphoma. Blood, The Journal of the American Society of Hematology. 2019 Mar 14;133(11):1257-9.
37. Enzan N, Kitadate A, Tanaka A, Matsue K. Incisional random skin biopsy, not punch biopsy, is an appropriate method for diagnosis of intravascular large B-cell lymphoma: a clinicopathological study of 25 patients. British Journal of Dermatology. 2019 Jul;181(1):200-1.
38. Cho HG, Sheu SL, Kuo KY, Ally MS, Bailey EE, Kim J, et al. Limited role of random skin biopsy in the diagnosis of intravascular lymphoma in adult patients with hemophagocytic lymphohistiocytosis. Acta Haematologica. 2017;138(1):33-8.
39. Voso MT, Ottone T, Lavorgna S, Venditti A, Maurillo L, Lo-Coco F, et al. MRD in AML: The role of new techniques. Frontiers in Oncology. 2019;9: 655.
40. Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. New England Journal of Medicine. 2017 Aug 3;377(5):454-64.
41. Herbreteau G, Vallée A, Charpentier S, Normanno N, Hofman P, Denis MG. Circulating free tumor DNA in non-small cell lung cancer (NSCLC): clinical application and future perspectives. Journal of Thoracic Disease. 2019 Jan;11(Suppl 1): S113-Ss126.
42. Bronkhorst AJ, Ungerer V, Holdenrieder S. The emerging role of cell-free DNA as a molecular marker for cancer management. Biomolecular detection and quantification. 2019 Mar 1;17: 100087.
43. Diehn M, Alizadeh AA, Adams HP, Lee JJ, Klassen S, Palma JF, et al. Early prediction of clinical outcomes in resected stage II and III colorectal cancer (CRC) through deep sequencing of circulating tumor DNA (ctDNA). Journal of Clinical Oncology 2017 35:15_suppl, 3591-3591
44. Kurtz DM, Scherer F, Jin MC, Soo J, Craig AF, Esfahani MS, et al. Circulating tumor DNA measurements as early outcome predictors in diffuse large B-cell lymphoma. Journal of Clinical Oncology. 2018 Oct 1;36(28):2845-53.
45. Huet S, Salles G. Potential of Circulating Tumor DNA for the Management of Patients with Lymphoma. JCO Oncology Practice. 2020 May: JOP-19. 00691.
46. Rossi D, Kurtz DM, Roschewski M, Cavalli F, Zucca E, Wilson WH. The development of liquid biopsy for research and clinical practice in lymphomas: Report of the 15-ICML workshop on ctDNA. Hematological Oncology. 2020 Feb;38(1):34-7.
47. Suehara Y, Sakata-Yanagimoto M, Hattori K, Nanmoku T, Itoh T, Kaji D, et al. Liquid biopsy for the identification of intravascular large B-cell lymphoma. Haematologica. 2018 Jun;103(6): e241-e244.
48. Schrader AM, Jansen PM, Willemze R, Vermeer MH, Cleton-Jansen AM, Somers SF, et al. High prevalence of MYD88 and CD79B mutations in intravascular large B-cell lymphoma. (1528-0020 (Electronic)).