Abstract
In the manuscript entitled “The ion channels and transporters gene expression profile indicates a shift in excitability and metabolisms during malignant progression of Follicular Lymphoma” [1], we reported recent advances in our understanding of how the gene expression profile of ion channels and transporters (ICT-GEP) contributes to identify specific signatures associated with Follicular Lymphoma (FL), with those FL that acquire chemoresistance after a relapsing-remitting course, and with the more aggressive Diffuse Large Cell Lymphoma (DLBCL), which may represent the evolution of FLs. From the analysis of how ICT-GEP changes during FL malignant progression emerged the progressive decrease in the expression of genes encoding those channels which are responsible for the maintenance of the ionic homeostasis and the driving force for Ca2+ entry, upon cell activation, as well as a shift from a glycolytic to an oxidative metabolism.
Commentary
Lymphoma represents the most common form of hematological malignancy in the developed world, accounting for 3.6% of all cancers and 55.6% of all blood cancers in Europe, with non-Hodgkin lymphomas (NHL) representing 90% of cases.
We have based our study on FL and DLBCL, which collectively account for nearly half of all NHLs [2,3].
FL is the commonest indolent NHL, a low-grade germinal centre-derived tumour characterized by the deregulation of several driver genes and signalling pathways involved in the pathogenesis [4-7]. It is generally considered incurable, in fact after a relapsing-remitting course, 10% of cases will not respond to chemo-immunotherapy. Furthermore, over the course of the illness, roughly half of the patients develop a more aggressive disease, which may eventually transform into a DLBCL [8,9].
DLBCL is thought to arise from the germinal centre (GC) stage of B cell differentiation, is the most aggressive lymphoma, despite being curable in 60-70% of patients with combined immunochemotherapy. From the analysis of GEP emerged the existence of at least 2 genetic subtypes of DLBCL that can be tracked to different stages of B-cell differentiation, resembling either germinal center B-cells (GCB) or activated B-cells (ABC) [10].
These tumors arise from the transformation of lymphocytes in a relatively late stage of development, which underwent the process of antigen receptors’ diversification. During this process, B cells repeatedly rearrange their DNA to produce a unique and functional receptor, undergo massive clonal expansion, and can then live as ‘memory’ cells for long times. These traits, fundamental for the response to infectious agents, at the same time render the cells vulnerable to neoplastic transformation.
A key initiating genetic event is in fact a chromosomal translocation that leads to the deregulation of the expression of specific driver genes. B cells are particularly susceptible to this kind of recombinatorial events because of the Class switch recombination process (CSR), where the constant regions of the B cell receptor are changed to modify the effector function of the antibody [11]. Thus, when translocation breakpoints are located in the switch regions of the heavy chain constant genes, it is likely the result of a mistaken CSR. In the considered lymphomas the regulation of specific driver genes is often altered, thus it is important to study the expression of different genes, characterizing their GEP. This is the case of the deregulation of BCL2 in FL and BCL6 in DLBCL, that follows oncogene traslocations [12] involving the immunoglobulin switch region.
Thus the characterization of the gene expression profile (GEP) and the identification of specific signatures is emerging as a novel tool for the classification of non- Hodgkin lymphoma, the identification of different molecular subtypes and the understanding of neoplastic B-cell biology [10]. This can lead to the diagnostic and prognostic characterisation of the disease and the development of new drugs specifically targeted to the different genetic subtypes of lymphomas.
Also chronic self-antigen-induced signalling through the B-cell receptor (BCR), a characteristic of normal B cells that have been exposed to antigen [13], is implicated in lymphomagenesis of mature B lymphocytes. Particularly in ABC DLBCL [13,14,15], a chronic BCR signalling, that might be both induced by an invading pathogen or selfantigen, is essential for the survival of the tumour cells.
Beside stimulation, another possibility to affect BCR signalling is perturbing the intracellular Ca2+ influx which follows the antigen binding. This can be altered by modulating the expression of the ICT responsible for the maintaining of the V rest and the driving force for Ca2+ entry such as Kv1.3 and KCa3.1 and others [16].
ICTs, by coupling extracellular events with intracellular responses and maintaining intracellular ionic homeostasis, are implicated in cancer establishment and progression. Thus, they are emerging as novel cancer biomarkers [17,18] for different cancers including gliomas, breast, and lung cancers [18-20]. Indeed, ICT detection and quantification have been proposed as a novel tool to predict survival and clinical outcome. Additionally, a role of a specific K+ channels, Kv 11.1, also known as hERG1, has recently been associated with chemoresistance and invasiveness of leukemic blasts [21,22].
The expression and role of ICTs, has been thoroughly investigated in T-cells since almost thirty years, leading to the uncontested role of different K+ and Ca2+ currents in the regulation of lymphocyte response to antigens and subsequent activation [16,23]. On the contrary, few studies have addressed the expression and role of ICTs in B-cell lymphomas, and all of them have been focused on the functional characterization of single molecular entities [10,24-26]. Overall, no lymphoma ICT-GEP was provided so far.
Based on these premises and with the aim to identify different profiles related to the progression of the disease and therefore their potential translational relevance, we have determined the ICT-GEP of FL, compared with the one of FL after the relapse, and with the more aggressive DLBCL.
The GEP was determined by cDNA microarray transcriptomic characterization of samples both from a cohort of patients specifically enrolled for the study (54 consecutive diseased lymph node samples, of which 11 diagnosed as FL and 2 as DLBCL passed an RNA integrity evaluation) and compared with that of normal lymph nodes from pooled healthy donors, as well as from public datasets.
Genes showing a fold change (log2 fold change) ≥ 2 (corrected p-value < 0.01) were considered deregulated. Applying such thresholds, 3988 DE genes, mostly under expressed, emerged, showing no relevant difference in the stratification within the 11 FL samples. This indicates a substantial homogeneity of the molecular characteristics of the samples, in agreement with the clinical and pathological homogeneity of the cohort analyzed.
Focusing on the ICT genes, 46 resulted deregulated, and 8 of them were confirmed DE also from the analysis of the GSE65135 FL dataset deposited into the GEO database. These 8 commonly DE ICT genes hence potentially represent the ICT gene expression signature of Follicular Lymphoma (Figure 1). Among them, SLC2A1 encoding the glucose transporter 1 (Glut1) and SLC9A9 (encoding the Na+/H+ antiporter NHE9) were over expressed in both datasets.
An upregulation of K+ channels, such as KCNN4 and KCNAB2, which encode for the alpha and beta subunits of Ca2+-dependent K+ channels, respectively, emerged. Notably, the Ca2+-dependent K+ channel KCa3.1 encoded by KCNN4, is one of the two K+ currents (together with Kv1.3) involved in lymphocyte activation and proliferation, and its expression marks the differentiation into activated naïve B cells and IgD+ CD27+ memory B cells [22]. This supports the notion FLs derive form germinal centre cells in an activated state.
Despite KCNN4 overexpression an overall reduced driving force for Ca2+ entry and effector potential is conceivable, being other potassium channel encoding gene, such as KCNH2 under expressed, differently from what occurs in other human cancers including leukemias [27].
The over expression of two solute carriers, SLC2A1 (encoding the glucose transporter member 1 (Glut-1) and SLC9A9 (encoding the Na+/H+ antiporter NHE9), merits further considerations. NHE proteins contribute to extrude the protons deriving from anaerobic metabolism determining the inverted pH characteristics of cancer cells [28], and Glut-1 is an indicator of the aerobic glycolysis, the main metabolic pathway of cancers (Warburg effect). In lymphomas, Glut-1 expression is apparently involved in FL transformation [29] and it is related to the NF- κB pathway, which turned out to be upregulated in FL from a Functional Annotation Analysis (FAA) applied to the identified DE-ICT. Also, the TNF pathway results upregulated in FL, and both of them are associated with the control of the metabolism [30,31].
Overall, the emerging ICT profile indicates that the main metabolic pathway of FL is represented by glycolysis, and that neoplastic B cells are able to respond to BCR stimulation, although at a lesser extent compared to activated normal B lymphocytes, being KCa3.1 the only relevant K+ channel identified. This channel might also contribute to tumor expansion, being known to sustain cell proliferation [16,23].
We then compared the DE genes in relapsed vs nonrelapsed FL patients and 38 DE ICT genes (16 over expressed and 22 under expressed) characterizing relapsed patients emerged. Notably among them, SLC2A1 and KCNN4 (encoding for KCa3.1 potassium channels) turned out to be under expressed, at difference from what occurs in the whole cohort of FL samples, where it resulted over expressed compared to normal lymphocytes (Figure 1).
This suggests that, upon relapse, neoplastic B cells become even less activated and undergo to down regulation of glycolysis. FL often acquire chemoresistance after relapse, and a shift to a fatty acid metabolism has been linked with resistance to chemotherapeutics in multiple cancer types [32]. This suggestion is also corroborated by the observation that ATPAF2, one of the factors involved in mitochondrial functioning [33], is over expressed in relapsed FL (log2 fold change value 5.52), and by the under expression of ATP9A. A depletion of ATP9A, has been associated to the retaining of Glut-1 in endosomes since it inhibits Glut-1 recycling, and hence reduces its expression on the cell surface [34].
Since 2-3% of relapsed FL can progress into a DLBCL [3,4], we also determined their specific ICT-GEP. To this purpose, we analysed the primary DLBCL samples present in our cohort, applying, the same analytical procedure previously described. 26 DE ICT genes emerged, seven of which (ANXA8, ATP9A, CACNA1E, CACNA1I, SLC26A1, SLC27A1, SLC7A4) did not overlap with the FL-ICT signature previously identified. This signature was further validated with the ICT GEP obtained from a publicly available dataset deposited into the GEO database (GSE12195) that contains microarray data from 71 DLBCL, and compared the GEP with that of centrocytes purified from the tonsils of 5 healthy subjects from the same dataset [35].
ATP9A, CACNA1I and SLC27A1 resulted commonly deregulated in both datasets, while did not match with the FL signature. This may hence represent a DLBCL-specific ICT signature (Figure 1).
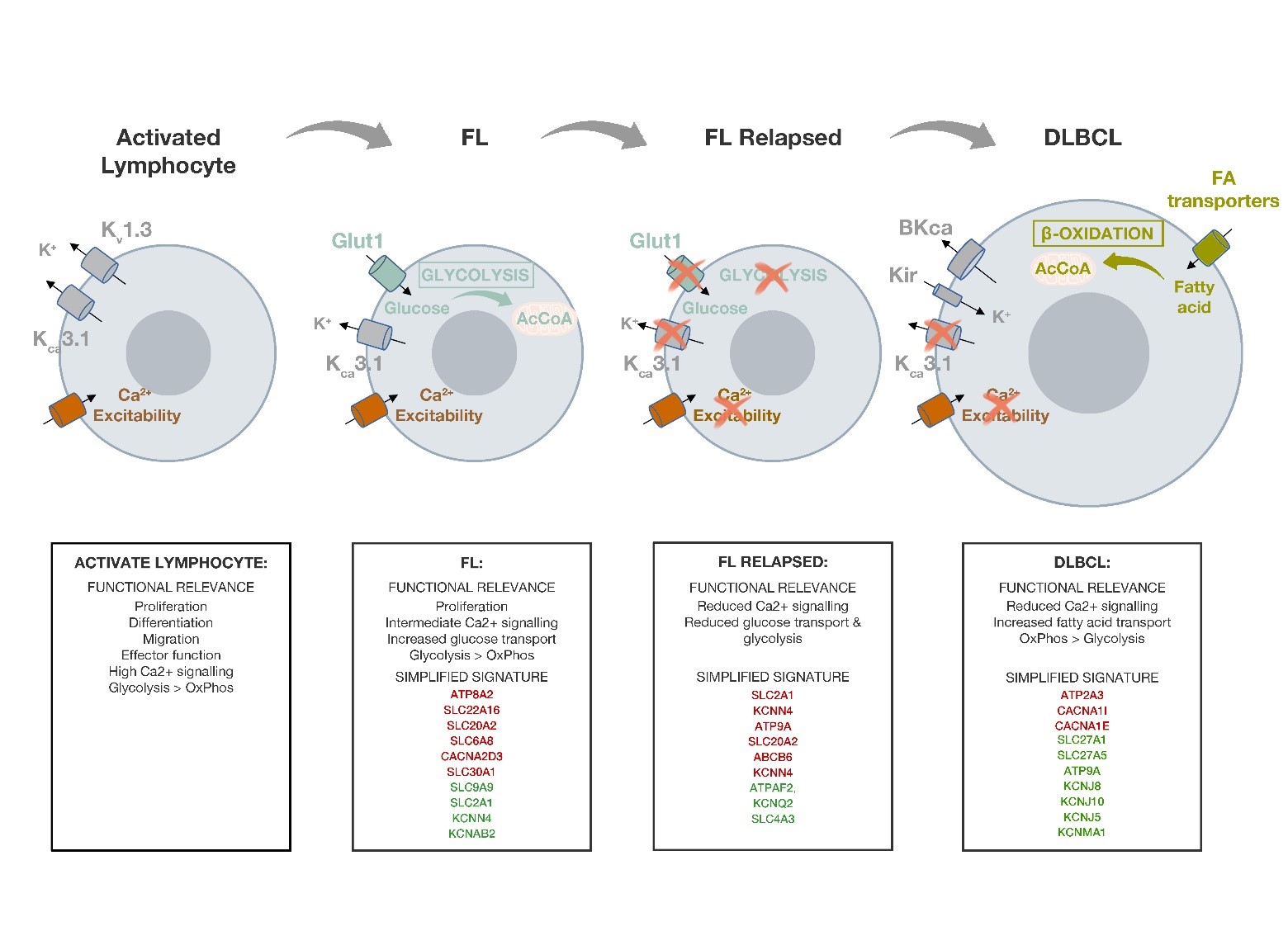
Figure 1. Summary figure indicating the changes in the expression of the different ICT and the associated metabolic pathways along lymphoma progression. For each lymphoma is reported a functional relevance summary of the main metabolic characteristic and of the Ca2+ response capacity and the identified ICT-DE signature of the different lymphomas described. In red are reported downregulated genes, in green the overexpressed ones.
From the identified DE ICT genes associated to DLBCL it is possible to draw some considerations:
The common dysregulation of ATP9A in both DLBCL and in the relapsed FL subgroup suggests its possible involvement in the progression of FL to the more aggressive DLBCL disease.
The changes in BCR induced Ca2+ response observed during the relapse of FL is now even more evident, in fact, CACNA1I (and the correlated CACNA1E) which encodes the alpha subunit of the low voltage-activated, T-type calcium channel, resulted down-regulated.
Such dysregulation of Ca2+ channel encoding genes is also accompanied by a drastic shift of the profile of K+ channels. In fact, the two K+ channels which mark normal B lymphocytes (KV1.3 and KCa3.1) are here substituted by the over expression of genes encoding inward rectifier K+ channels (Kir) and Big KCa channels (BK) (KCNJ8, KCNJ10, KCNJ5, KCNMA1)
In DLBCL is confirmed the reduction in the expression of genes involved in glycolysis, which we observed in relapsed FL, and it is corroborated by the up-regulation of the SLC27A1 fatty acid transporter. This transporter is responsible for the uptake of fatty acids for further beta oxidation [36] and suggests a shift from a glycolytic to an oxidative metabolic profile in DLBCL.
SLC27A1, by contributing to the supply of fatty acids from the surrounding adipocytes, and providing an alternative pathway for the metabolsim of acetyl CoA in the TCA cycle when cancer cells switch from a glycolytic to an oxidative metabolism, is also associated with tumorigenesis [37]. Thus, the downregulation of Glut-1 and the upregulation of the fatty acid transporter SLC27A1 in DLBCL indicate a shift from a glycolytic to an oxidative metabolism during lymphoma progression. In agreement, oxidative enzymes (ACAD10, ACAD8, ECHS1, HADHB) turned out to be over expressed (log2FC > 0) in DLBCL
Consistently, the nuclear corepressor-encoding gene NCoR1, whose down-regulation has been reported to drive the switch towards an oxidative metabolism [38], results under expressed in DLBCL.
In agreement with our data, an OxPhos subset of DLBCL, identified by a lower expression of NCoR1, has been recently described in [39,40]. Patients belonging to this subset might benefit from treatments perturbing the fatty acid oxidation program [39].
In conclusion the commented manuscript identifies for the first time an ICT signature of FL and its variations during the relapse after treatment and the progression towards DLBCL. From the analysis of the emerged deregulated ICT genes a change in lymphocytes response capacity and metabolism during the progression of FL emerged. The DE-ICT profile in relapsed FL patients, shows a decrease in glycolysis, which precedes the already described shift towards an oxidative metabolism in DLBCL, and a decrease in “activation” in terms of Ca2+ signalling in response to BCR stimulation. Such a suggestion is corroborated by a recent study showing lower intracellular Ca2+ levels following the blockade of Erg1 K+ channel [41].
This suggests that SLC27A1, and the OxPhos pathway, might constitute promising therapeutic targets, however a validation on different cohorts and detailed functional studies must be conducted to confirm the therapeutic potential of perturbing the fatty acid oxidative pathway in aggressive lymphomas.
In particular, the hints emerged should be deepened by further studies such as the electrophysiological characterization of the ionic currents determined by the identified DE genes, as well as the identification and evaluation of specific channel blockers. Moreover, it will be interesting to evaluate the signalling pathways and the Ca2+ entry in response to BCR stimulation, and further characterize the metabolic profile of the different lymphoma subtypes. This will open the way to identify and analyse the effects of drugs targeting fatty acid transporters and perturbing the OxPhos pathway in lymphomas, for a novel therapeutic approach.
Therefore, these findings might have a translational relevance through the identification of novel ICT and metabolism related therapeutic targets that might also lead to overcome the chemoresistance which characterizes relapsed FL.
Acknowledgment
This work was supported by grants from the Associazione Genitori contro le Leucemie e Tumori Infantili “Noi per Voi” and the Associazione Italiana per la Ricerca sul Cancro (AIRC).
The data discussed in this publication have been deposited in NCBI’s Gene xpression Omnibus (Magi, A. et al. 2019) and are accessible through GEO Series accession number GSE 126247 (https://www.ncbi.nlm.nih.gov/geo/query/ acc.cgi?acc=GSE126247).
References
2. Novelli S, Briones J, Sierra J. Epidemiology of lymphoid malignancies: last decade update. Springerplus. 2013 Dec 1;2(1):70.
3. Morton LM, Wang SS, Devesa SS, Hartge P, Weisenburger DD, Linet MS. Lymphoma incidence patterns by WHO subtype in the United States, 1992-2001. Blood. 2006 Jan 1;107(1):265-276.
4. Glas AM, Kersten MJ, Delahaye LJ, Witteveen AT, Kibbelaar RE, Velds A, et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood. 2005 Jan 1;105(1):301-307.
5. Glas AM, Knoops L, Delahaye L, Kersten MJ, Kibbelaar RE, Wessels LA, et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. . Journal of Clinical. Oncology. 25, 390–398 (2007).
6. Schwaenen C, Viardot A, Berger H, Barth TF, Bentink S, Döhner H, et al. Microarray-based genomic profiling reveals novel genomic aberrations in follicular lymphoma which associate with patient survival and gene expression status. Genes, Chromosomes and Cancer. 2009 Jan;48(1):39-54.
7. Husson H, Carideo EG, Neuberg D, Schultze J, Munoz O, Marks PW, et al. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood, The Journal of the American Society of Hematology. 2002 Jan 1;99(1):282-289.
8. Swenson WT, Wooldridge JE, Lynch CF, Forman- Hoffman VL, Chrischilles E, Link BK. Improved survival of follicular lymphoma patients in the United States. Journal of Clinical Oncology. 2005 Aug 1;23(22):5019-5026.
9. Irish JM, Myklebust JH, Alizadeh AA, Houot R, Sharman JP, Czerwinski DK, et al. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proceedings of the National Academy of Sciences. 2010 Jul 20;107(29):12747-12754.
10. Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000 Feb;403(6769):503-511.
11. Küppers R, Dalla-Favera R. Mechanisms of chromosomal translocations in B cell lymphomas. Oncogene. 2001 Sep;20(40):5580-5594.
12. Baron BW, Nucifora G, McCabe N, Espinosa RI, Le Beau MM, McKeithan TW. Identification of the gene associated with the recurring chromosomal translocations t (3; 14)(q27; q32) and t (3; 22)(q27; q11) in B-cell lymphomas. Proceedings of the National Academy of Sciences. 1993 Jun 1;90(11):5262-5266.
13. Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010 Jan;463(7277):88-92.
14. Young RM, Wu T, Schmitz R, Dawood M, Xiao W, Phelan JD, et al. Survival of human lymphoma cells requires B-cell receptor engagement by self-antigens. Proceedings of the National Academy of Sciences. 2015 Nov 3;112(44):13447-13454.
15. Malcolm TI, Hodson DJ, Macintyre EA, Turner SD. Challenging perspectives on the cellular origins of lymphoma. Open Biology. 2016 Sep 1;6(9):160232.
16. Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annual Review of Immunology. 2015 Mar 21;33:291-353.
17. Lastraioli E, Iorio J, Arcangeli A. Ion channel expression as promising cancer biomarker. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2015 Oct 1;1848(10):2685-2702.
18. Ko JH, Ko EA, Gu W, Lim I, Bang H, Zhou T. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Molecular Cancer. 2013 Dec 1;12(1):106.
19. Ko JH, Gu W, Lim I, Bang H, Ko EA, Zhou T. Ion channel gene expression in lung adenocarcinoma: potential role in prognosis and diagnosis. PloS One. 2014 Jan 23;9(1):e86569.
20. Wang R, Gurguis CI, Gu W, Ko EA, Lim I, Bang H, et al. Ion channel gene expression predicts survival in glioma patients. Scientific Reports. 2015 Aug 3;5:11593.
21. Lima TI, Valentim RR, Araújo HN, Oliveira AG, Favero BC, Menezes ES, et al. Role of NCoR1 in mitochondrial function and energy metabolism. Cell Biology International. 2018 Jun;42(6):734-741.
22. Pillozzi S, Brizzi MF, Bernabei PA, Bartolozzi B, Caporale R, Basile V, et al. VEGFR-1 (FLT-1), β1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia: role in cell migration and clinical outcome. Blood. 2007 Aug 15;110(4):1238-50.
23. Arcangeli A, Pillozzi S, Becchetti A. Targeting ion channels in leukemias: a new challenge for treatment. Current Medicinal Chemistry. 2012 Feb 1;19(5):683-696.
24. Suguro M, Tagawa H, Kagami Y, Okamoto M, Ohshima K, Shiku H, et al. Expression profiling analysis of the CD5+ diffuse large B-cell lymphoma subgroup: development of a CD5 signature. Cancer Science. 2006 Sep;97(9):868-874.
25. Overes IM, De Rijke B, Van Horssen-Zoetbrood A, Fredrix H, De Graaf AO, Jansen JH, et al. Expression of P2X5 in lymphoid malignancies results in LRH-1- specific cytotoxic T-cell-mediated lysis. British Journal of Haematology. 2008 Jun;141(6):799-807.
26. Birerdinc A, Nohelty E, Marakhonov A, Manyam G, Panov I, Coon S, et al. Pro-apoptotic and antiproliferative activity of human KCNRG, a putative tumor suppressor in 13q14 region. Tumor Biology. 2010 Jan 1;31(1):33-45.
27. Pillozzi S, Brizzi MF, Balzi M, Crociani O, Cherubini A, Guasti L, et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia. 2002;16(9):1791-98.
28. NSTEIN SG, COHEN S. Role ofMembrane Potential and Volume-activated Na’/H+ Exchange. The Journal of General Physiology. 1987;89 185-213.
29. Sommermann TG, O’Neill K, Plas DR, Cahir- McFarland E. IKKβ and NF-κB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Research. 2011 Dec 1;71(23):7291-7300.
30. Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, et al. NF-kB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nature Cell Biology. 2011 Oct;13(10):1272-1279.
31. Samavati L, Lee I, Mathes I, Lottspeich F, Hüttemann M. Tumor necrosis factor α inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. Journal of Biological Chemistry. 2008 Jul 25;283(30):21134-21144.
32. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death & Disease. 2013 Mar;4(3):e532.
33. Wang ZG, White PS, Ackerman SH. Atp11p and Atp12p are assembly factors for the F1-ATPase in human mitochondria. Journal of Biological Chemistry. 2001 Aug 17;276(33):30773-30778.
34. Tanaka Y, Ono N, Shima T, Tanaka G, Katoh Y, Nakayama K, et al. The phospholipid flippase ATP9A is required for the recycling pathway from the endosomes to the plasma membrane. Molecular Biology of the Cell. 2016 Dec 1;27(24):3883-3893.
35. Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kB in diffuse large B-cell lymphoma. Nature. 2009 Jun;459(7247):717-721.
36. Nath A, Chan C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Scientific Reports. 2016 Jan 4;6:18669.
37. Navarro P, Bueno MJ, Zagorac I, Mondejar T, Sanchez J, Mourón S, et al. Targeting tumor mitochondrial metabolism overcomes resistance to antiangiogenics. Cell Reports. 2016 Jun 21;15(12):2705-2718.
38. Lima TI, Valentim RR, Araújo HN, Oliveira AG, Favero BC, Menezes ES, et al. Role of NCoR1 in mitochondrial function and energy metabolism. Cell Biology International. 2018 Jun;42(6):734-41.
39. Caro P, Kishan AU, Norberg E, Stanley IA, Chapuy B, Ficarro SB, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012 Oct 16;22(4):547-560.
40. Maneix L, Catic A. Touch and go: nuclear proteolysis in the regulation of metabolic genes and cancer. FEBS letters. 2016 Apr 1;590(7):908-923.
41. Manoli S, Coppola S, Duranti C, Lulli M, Magni L, Kuppalu N, et al. The Activity of Kv 11.1 Potassium Channel Modulates F-Actin Organization During Cell Migration of Pancreatic Ductal Adenocarcinoma Cells. Cancers. 2019 Jan 23;11(2):135.