Commentary
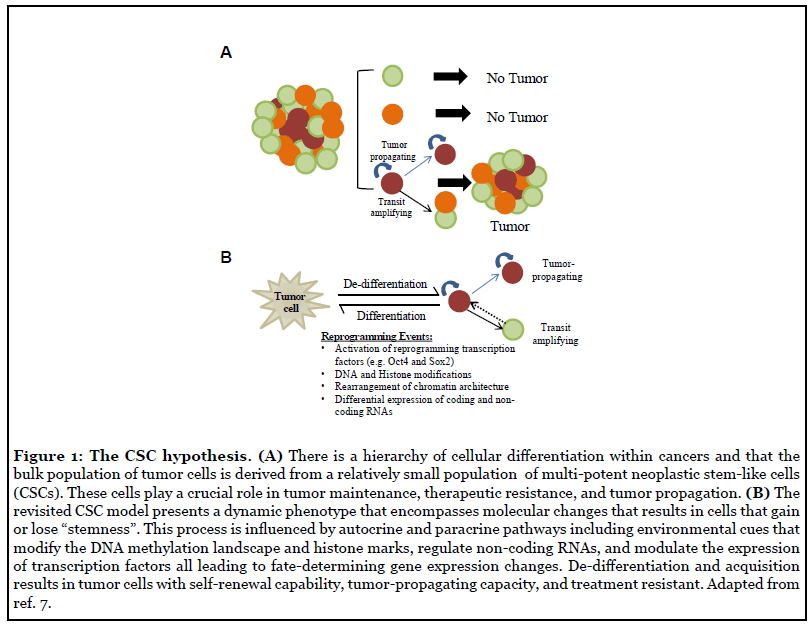
If cancer is considered the emperor of all maladies, then it is fair to say that cancer stem cells (CSCs) are the seeds that spread this evil. Since the late 1800s, pathologists have increasingly documented the cellular heterogeneity within solid tumors and recognized that some tumor cells seemed less differentiated than others [1]. In the late 1930s Furth and Kahn showed that a single cell was sufficient to propagate tumor xenografts that recapitulate with Conversely, following in Furth and Kahn’s footsteps high fidelity the heterogeneity of clinical tumors [2-4]., Pierce showed that undifferentiated teratocarcinoma cells are highly tumorigenic, however, they lose tumor-propagation capacity upon differentiation [5]. In pioneering lineage tracing experiments, Pierce also showed that labeled undifferentiated tumor cells gave rise to fully differentiated cells, and surprisingly, the labeled differentiated cell progeny lost tumor-propagating capacity [6]. This seminal work gave birth to what we recognize today as the Cancer Stem Cell Hypothesis. This theory postulates that there is a hierarchy of cellular differentiation within tumors, similar to the cellular hierarchies that exist during normal tissue development, and only a small subset of multi-potent neoplastic stem-like cells can propagate tumors through asymmetric cell division [7] (Figure 1A). The clinical implication of this cellular hierarchy is that eradicating the apical tumor-progenitor cell (i.e. CSC) will more effectively treat tumors and prevent recurrence.
Highly aggressive tumors display a poorly differentiated cell phenotype characterized by high expression of genes enriched in pluripotent embryonic stem cells (ESCs) [8,9]. Expression of a defined set of transcription factors, the so-called Yamanka [10] factors (i.e. Oct4, Sox2, Klf4, and c-Myc), is sufficient to reprogram mouse and human cells to an induced-pluripotent state. These iPS cells resemble embryonic stem cells since they possess the capacity to differentiate into all tissue sub-types [10]. In cancer, expression of these transcription factors (Oct4, Sox2, c-Myc, and Klf4) has been found to correlate with poor prognosis and tumor progression [11]. The high similarity in gene expression profiles of embryonic stem cells (ESCs) and high-grade tumors [8] further supports the molecular parallels between the stem cell phenotype, induced pluripotency, and cancer [8]. This suggests a de-differentiation mechanism whereby expression and function of reprogramming transcription factors influence the tumorigenic potential of cells by driving them to less differentiated and potentially more aggressive stem-like states.
Neoplastic cells with multi-potent stem cell properties have now been identified in most cancer types and are largely recognized as the tumor-propagating cell population [12,13]. In fact, lineage-tracing experiments show that stem-like cells are responsible for tumor re-growth after therapy [14] and this process is in part mediated by known drivers of stemness [15]. Stem-like cell subpopulations within tumors are endowed with the capacity to efficiently propagate tumor xenografts with high histopathologic fidelity, self-renew through asymmetric cell division and generate transit amplifying progenitor cells with limited self-renewing capacity [16,17]. Mounting evidence has led to the paradigm-shifting realization that non-stemlike cancer cells can acquired stem-like traits depending on cell-intrinsic and extrinsic factors, including current therapeutics designed to get rid of the tumor itself [18-20]. This ability to switch phenotypes, commonly referred to as plasticity, is a key feature of tumor cells that allows them to undergo de-differentiation to maintain tumor growth, transition to a drug-resistant state, create intratumor cellular heterogeneity, and drive tumor recurrence [21]. Tumor cell plasticity is driven, in large part, by pluripotency factors that coordinate reprogramming molecular circuits that function within the context of an aberrant cancer genome [7,20]. This plasticity requires extensive transcriptional changes coordinated, to a large extent, by epigenetic alterations including nucleosome re-positioning and re-distribution of DNA and histone modifications [23-25]. Not surprisingly, increasing data shows that epigenetic reprogramming and tumor cell dedifferentiation to a stem-like phenotype are key events determining acquisition of therapy-resistance and tumorpropagating capacity in cancer cells [26,27]. These newfound mechanisms have forced a significant revision of the original strictly hierarchical model and current iterations of the CSC hypothesis incorporate the inherent plasticity of malignant cells and their capacity to dynamically de-differentiate to generate cells with tumor-propagating capacity [7,20] (Figure 1B).
We now understand that CSCs play critical roles in driving intra-tumor cellular heterogeneity by establishing dynamic cellular transitions within tumors [28-30]. Cells within the same tumor can display distinct phenotypes that affect their growth rate, survival, migration, therapyresponse, and tumor-propagating capacity [28,31-34]. For instance, in GBM, transcriptome analysis of clinical samples identified distinct subtypes based on molecular signatures (i.e. mesenchymal, classical, and proneural) [35] with the mesenchymal subtype, thought to be the most aggressive [35,36], sharing a high-degree of similarity with stem cells at the transcriptome and epigenetic level [37]. Moreover, single-cell analysis shows that GBM cells exist in at least four different cellular states and can transition dynamically between molecular subtypes depending on cell-intrinsic and extrinsic factors [38,39]. This highdegree of transcriptional and epigenetic adaptability is thought to influence the emergence of therapy-resistant GBM cell sub-populations [26] and plays a pivotal role in establishing intra-tumor heterogeneity [40]. Interestingly, reprogramming transcription factors Oct4 and Sox2 are reported to play a driving role in the mesenchymal transition in multiple neoplasms including oral squamous carcinomas, nasopharyngeal carcinomas, and breast cancer [41-44].
Over the last decade our lab has been focused on understanding what drives the de-differentiation of GBM cells to their tumor-propagating phenotype with the goal of identifying and exploiting tumor cell vulnerabilities. We were at the forefront of identifying a role for the Yamanka [10] factors in controlling the tumor-propagating phenotype of GBM stem-like cells by oncogenic c-Met signaling [45-48]. We showed that c-Met is activated and functional in glioblastoma (GBM) neurospheres enriched for glioblastoma tumor-initiating stem cells and that c-Met expression/function correlates with stem cell marker expression and the neoplastic stem cell phenotype of GBM cells. c-Met activation was found to induce the expression of reprogramming transcription factors known to support embryonic stem cells and induce differentiated cells to form pluripotent stem (iPS) cells, and c-Met activation inhibited the effects of forced differentiation in glioblastoma neurospheres. We further demonstrated that reprogramming transcription factor Nanog mediated the ability of c-Met to induce the stem cell phenotype of GBM cells [46]. These in vitro observations lead us to examine the effects of in vivo c-Met pathway inhibitor therapy on tumor-propagating stem-like cells in human GBM xenografts. We demonstrated that c-Met pathway inhibition via neutralizing anti-hepatocyte growth factor (HGF) monoclonal antibody L2G7 or with the c-Met kinase inhibitor PF2341066 (Crizotinib) reduced tumor growth, depleted tumors of sphere-forming cells, and inhibited tumor expression of stem cell markers CD133, Sox2, Nanog, and Musashi [48]. Simultaneous findings by DeBacco et.al. show that expression of c-MET associates with neurospheres expressing the gene signature of mesenchymal and proneural subtypes and functions as a marker of glioblastoma stem cells [45]. Contemporaneous results started to paint the picture that the tumor cell microenvironment plays a fundamental role in establishing and maintaining the aforementioned cellular plasticity. The hypoxic environment [49], juxtracrine/paracrine signaling [50], the perivascular niche [51,52], all work together to establish bi-directional signaling cascades to support, and in some cases, drive the stem cell phenotype of GBM cells. Taken together, these findings established dynamically-regulated de-differentiation mechanisms involved in cancer stem-cell generation and maintenance in GBM.
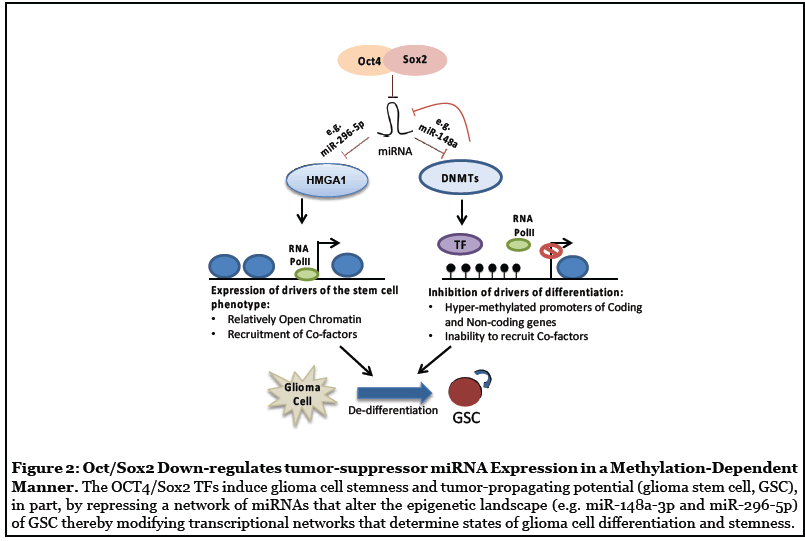
In follow-up novel and original studies, we uncovered a molecular circuit in which Oct4/Sox2 drives GSC cellular transitions and tumor propagation, in part, by repressing miRNAs that regulate two distinct epigenetic mechanisms: (1) changes in chromatin architecture through the action of HMGA1, and (2) DNMT-dependent DNA methylation events (Figure 2). We found that Oct4 and Sox2 expression levels strongly correlate with expression of the DNMT1 and DNMT3B in primary GBM neurosphere isolates and that Oct4 and Sox2 directly transactivate Dnmt genes and induce glioma cell hyper-methylation. Moreover, DNMTs are enriched in GBM stem-like cell subsets and that pharmacological inhibition of DNMTs inhibits the capacity of GBM stem cells to self-renew as neurospheres [19]. Additionally, Oct4 and Sox2 silence a subset of miRNAs in GBM stem-like cells via a mechanism involving DNMT up-regulation and promoter hyper-methylation and identified miR-148a-3p as a downstream effector of this network [19]. This study established a molecular circuit by which the core reprogramming transcription factors Oct4 and Sox2 regulate the GBM stem-like phenotype by modifying epigenetic networks. In a parallel study, we identified HMGA1, a remodeler of chromatin architecture, as a functional target of miR-296-5p and an intermediary by which miR-296-5p regulates Sox2 expression [25]. We found that HMGA1 is induced as a result of miR-296-5p repression by Oct4 and Sox2 co-expression and functions to induce reprogramming signals in GBM cells required for maintaining the stem cell phenotype [25]. Furthermore, we identified a mechanism by which HMGA1 associates with and displaces Histone H1 from the Sox2 promoter and in doing so induces Sox2 expression [25]. Importantly, we found that normalization of epigenetic networks by reconstituting either miR-148a-3p or miR-296-5p inhibits the tumor propagating capacity of GBM stem cells [19,25].
Despite major advances in our understanding of cancer at the molecular level, mechanism-based treatment modalities remain limited [53]. Non-coding RNAs, in particular miRNAs, are emerging as critical regulators of cell fate and oncogenesis [54,55]. miRNAs selectively inhibit gene expression primarily by targeting mRNA for degradation usually via complementary 3’-UTR seed sequences [55]. Numerous miRNAs have been found to regulate tumorigenesis and cancer cell stemness by targeting tumor-suppressing or tumor promoting transcripts [56,57]. Activation of tumorigenic cascades involve dysregulation of multi-dimensional molecular networks with miRNAs playing key roles in the process [19,25,57]. Our work and that of others suggests that reconstituting tumor-suppressive miRNAs or inhibiting oncogenic miRNAs can normalize these dysregulated molecular networks, inhibit tumor growth and enhance the effects of current standards-of-care [55,58,59].
One of the biggest challenges for RNAi-based therapeutic strategies is their stability, delivery and bioavailability [60]. Two main approaches have been at the forefront of tackling the problem of RNA instability: stabilization by chemically modifying the RNA oligonucleotides or the use of delivery vehicles to protect the RNA molecules until it reaches the desired site of action [55]. Both strategies have yielded success, however recent advances in nanomedicine are tilting the balance towards the use of nanocarriers to deliver the RNAi cargo as a more flexible and advantageous mode of delivery [61]. A powerful advantage of these nextgeneration nanocarriers is that they readily accommodate payloads consisting of multiple miRNAs and/or miRNA inhibitors (i.e. antagomirs), and thus offer an ideal vehicle for implementing multi-miRNA normalization strategies as proposed above [59,62]. These types of carriers can be optimized for siRNA/miRNA delivery to specific cell types, providing an extra level of control to minimize off-target effects [63,64].
To translate the concept of miRNA network normalization, we developed and characterized novel bioreducible poly (β-amino ester) (PBAE) polymers that can effectively deliver miRNA mimics and/or inhibitors (i.e. antagomirs) to inhibit the GBM stem cell phenotype in vitro [59]. A powerful advantage of PBAE nanoparticles is that they readily accommodate payloads consisting of multiple miRNAs, both mimics and antagomirs, and thus offer an ideal vehicle for implementing such multi-miRNA normalization strategies [59,62]. In fact, we found that PBAE/miRNA nanoparticles have a high loading capacity allowing for facile codelivery of nanoparticles containing multiple miRNA types to cells of interest (e.g. miR-148a- 3p and miR-296-5p) [59]. Using a technique that closely resembles clinically translatable convection enhanced delivery (CED) [59] we show, for the first time, that nanoparticles containing miR-148a-3p and miR-296-5p penetrate an established tumor in vivo, inhibit the growth of established GBM xenografts and prolong survival with actual cures in mouse models [59]. Building upon these mechanistic and conceptual insights we recently identified miR-486-5p as a Sox2-induced onco-miRNA that regulates GBM stem-like cells by targeting tumor suppressor genes PTEN and FoxO1 [65]. Inhibition of endogenous miR-486- 5p induced cell death by upregulating proapoptotic protein BIM via a PTEN-dependent mechanism [65] and delivery of miR-486-5p antagomirs to pre-established orthotopic GBM neurosphere-derived xenografts using advanced nanoparticle formulations inhibited tumor growth and enhanced the cytotoxic response to ionizing radiation [65].
Despite the promise of targeted cancer therapies, their clinical success against solid tumors has been limited due to resistant cell sub-populations [66]. These subpopulations of cancer cells escape therapy by (i) mutations in the target, (ii) reactivation of the targeted pathway, (iii) activating alternative pathways, or (iv) transitioning to a resistant state (e.g. stem-like state – see Figure 1B). Therefore, the use of combinatorial therapies to target parallel oncogenic pathways that may lead to a resistant phenotype is favored to tackle cellular heterogeneity and inhibit cellular transitions. Understanding the molecular mechanism that drive the stem-like state of cancer cells and efficiently targeting them will yield therapeutics that both reduce cellular heterogeneity and block bi-directional cellular transitions (Figure 1). One feature of miRNAbased therapies, the advantages of which are frequently overlooked, is the ability of one miRNA to target multiple mRNA transcripts [67,68]. This promiscuous quality of miRNAs is often perceived as a source of concern for therapeutic development [55]. However, carefully selecting one miRNA to target multiple mRNAs dysregulated during tumorigenesis can allow the targeting of multiple parallel oncogenic pathways using a single agent, enhancing therapeutic efficacy and reducing chances of tumor recurrence.
Our work highlights how novel developments in mechanisms of cell fate regulation can combine with nanomedicine to provide new avenues to develop innovative pre-clinical molecular therapeutics [69] (Figure 3). However, to translate our pre-clinical success to patients we must overcome some significant obstacles involving safe and efficacious delivery of these nextgeneration molecular therapeutics. Ideally, a minimally invasive procedure consisting of systemic delivery of our nano-miR therapy would be preferred. To achieve this goal, improvement in polymer stability and blood-brainbarrier (BBB) penetration are a priority. We envision the next-generation of polymers used to deliver stemcell inhibiting miRNAs having a longer half-life and chemical modifications to enhance BBB penetration to facilitate systemic delivery [70]. Alternatively, advances in ultrasound technology now allow for localized, transient disruption of the BBB [71]. We can envision using this technology to permeabilize regions in the brain to enhance uptake of nanoparticles and specifically target tumor regions. Additionally, breakthroughs in the field of cranioplasty surgery now allows for next-generation implants harboring technology such as shunts to monitor and control intracranial pressure [72]. It shouldn’t be surprising if in the near future this technology is to create devices for ambulatory CED uses that will perfectly compliment drug delivery application such as our preclinical therapeutics.
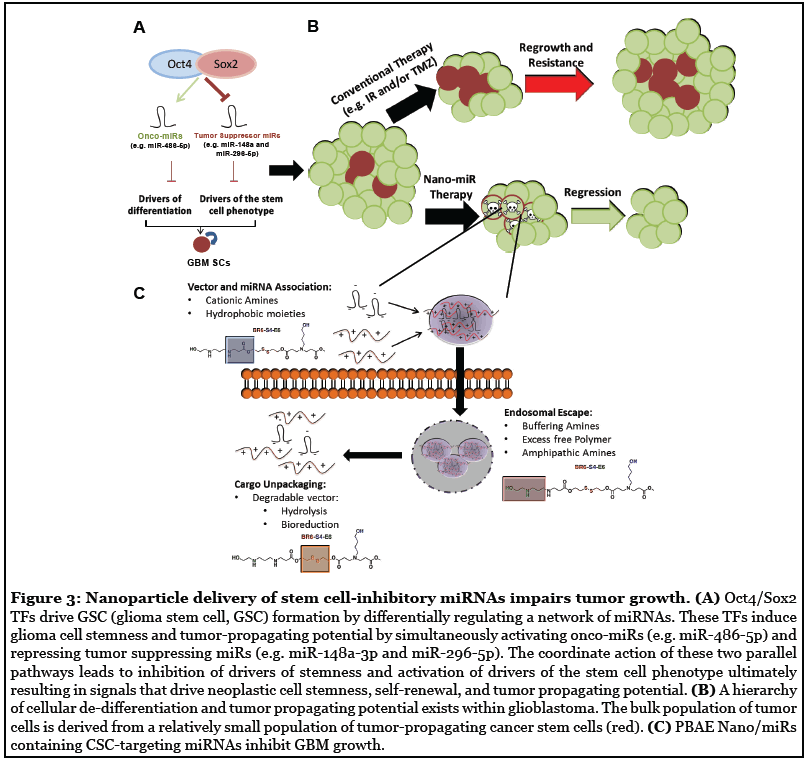
Intra-tumor heterogeneity presents one of the biggest challenges in the development of solid cancer therapeutics [40]. We now understand CSCs play critical roles in driving intra-tumor cellular heterogeneity by establishing dynamic cellular transitions within tumors [28-30]. We are entering an exciting time where our knowledge of the molecular drivers of cancer plasticity combined with innovations in nanomedicine and engineering are paving the road for the next generation of rational molecular therapeutics. These multidisciplinary efforts are certain to bring us closer to uprooting the seed of all evil to significantly impact cancer patient outcomes.
References
1. DeVita Jr VT, Rosenberg SA. Two hundred years of cancer research. New England Journal of Medicine. 2012 Jun 7;366(23):2207-14.
2. Furth J, Kahn MC, Breedis C. The transmission of leukemia of mice with a single cell. The American Journal of Cancer. 1937 Oct 1;31(2):276-82.
3. Hewitt HB. Studies of the dissemination and quantitative transplantation of a lymphocytic leukaemia of CBA mice. British journal of cancer. 1958 Sep;12(3):378- 401.
4. Makino S. Further evidence favoring the concept of the stem cell in ascites tumors of rats. Annals of the New York Academy of Sciences. 1956 Mar;63(5):818-30.
5. Kleinsmith LJ, Pierce GB. Multipotentiality of single embryonal carcinoma cells. Cancer Research. 1964 Oct 1;24(9):1544-51.
6. Pierce GB, Wallace C. Differentiation of malignant to benign cells. Cancer research. 1971 Feb 1;31(2):127-34.
7. Lopez-Bertoni H, Li Y, Laterra J. Cancer stem cells: dynamic entities in an ever-evolving paradigm. Biology and medicine (Aligarh). 2015 Nov;7(Suppl 2):001.
8. Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A,et al. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nature genetics. 2008 May;40(5):499.
9. Brown DV, Filiz G, Daniel PM, Hollande F, Dworkin S, Amiridis S, et al. Expression of CD133 and CD44 in glioblastoma stem cells correlates with cell proliferation, phenotype stability and intra-tumor heterogeneity. PLoS One. 2017 Feb 27;12(2):e0172791.
10. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell. 2006 Aug 25;126(4):663- 76.
11. Friedmann-Morvinski D, Verma IM. Dedifferentiation and reprogramming: origins of cancer stem cells. EMBO reports. 2014 Mar;15(3):244-53.
12. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nature Reviews Cancer. 2012 Nov;12(11):767-75.
13. Lathia J, Liu H, Matei D. The clinical impact of Cancer stem cells. The Oncologist.2019 Dec 17;.2019-0517.
14. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012 Aug;488(7412):522-6.
15. Gangemi RM, Griffero F, Marubbi D, Perera M, Capra MC, Malatesta P, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009 Jan;27(1):40-8.
16. Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences. 2003 Apr 1;100(7):3983-8.
17. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. nature. 2004 Nov;432(7015):396-401.
18. Auffinger B, Tobias AL, Han Y, Lee G, Guo D, Dey M, et al. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death & Differentiation. 2014 Jul;21(7):1119-31.
19. Lopez-Bertoni H, Lal B, Li A, Caplan M, Guerrero- Cazares H, Eberhart CG, et al. DNMT-dependent suppression of microRNA regulates the induction of GBM tumor-propagating phenotype by Oct4 and Sox2. Oncogene. 2015 Jul;34(30):3994-4004.
20. Liu Y, Yang M, Luo J, Zhou H. Radiotherapy targeting cancer stem cells “awakens” them to induce tumour relapse and metastasis in oral cancer. International Journal of Oral Science. 2020 Jun 24;12(1):1-2
21. Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nature Reviews Drug Discovery. 2020 Jan;19(1):39-56.
22. Iglesias JM, Gumuzio J, Martin AG. Linking pluripotency reprogramming and cancer. Stem Cells Translational Medicine. 2017 Feb;6(2):335-9.
23. Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nature Reviews Cancer. 2011 Oct;11(10):726-34.
24. Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Molecular Oncology. 2012 Dec 1;6(6):620-36.
25. Lopez-Bertoni H, Lal B, Michelson N, Guerrero- Cazares H, Quinones-Hinojosa A, Li Y, et al. Epigenetic modulation of a miR-296-5p: HMGA1 axis regulates Sox2 expression and glioblastoma stem cells. Oncogene. 2016 Sep;35(37):4903-13.
26. Liau BB, Sievers C, Donohue LK, Gillespie SM, Flavahan WA, Miller TE, et al. Adaptive chromatin remodeling drives glioblastoma stem cell plasticity and drug tolerance. Cell Stem Cell. 2017 Feb 2;20(2):233-46.
27. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010 Apr 2;141(1):69-80.
28. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl j Med. 2012 Mar 8;366:883-92.
29. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013 Sep;501(7467):328-37.
30. Sottoriva A, Verhoeff JJ, Borovski T, McWeeney SK, Naumov L, Medema JP, et al. Cancer stem cell tumor model reveals invasive morphology and increased phenotypical heterogeneity. Cancer Research. 2010 Jan 1;70(1):46-56.
31. Yung WK, Shapiro JR, Shapiro WR. Heterogeneous chemosensitivities of subpopulations of human glioma cells in culture. Cancer research. 1982 Mar 1;42(3):992-8.
32. Hyo-eun CB, Ruddy DA, Radhakrishna VK, Caushi JX, Zhao R, Hims MM, et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nature medicine. 2015 May;21(5):440-8.
33. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011 Apr;472(7341):90.
34. Verhaak RG. Cancer Genome Atlas Research Network: Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell. 2010;17:98- 110.
35. Behnan J, Finocchiaro G, Hanna G. The landscape of the mesenchymal signature in brain tumours. Brain. 2019 Apr 1;142(4):847-66.
36. Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, Weinstein JN, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell. 2018 Apr 5;173(2):338-54.
37. Neftel C, Laffy J, Filbin MG, Hara T, Shore ME, Rahme GJ, et al. An integrative model of cellular states, plasticity, and genetics for glioblastoma. Cell. 2019 Aug 8;178(4):835-49.
38. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014 Jun 20;344(6190):1396-401.
39. Nowell PC. The clonal evolution of tumor cell populations. Science. 1976 Oct 1;194(4260):23-8.
40. Vijayakumar G, Narwal A, Kamboj M, Sen R. Association of SOX2, OCT4 and WNT5A Expression in Oral Epithelial Dysplasia and Oral Squamous Cell Carcinoma: An Immunohistochemical Study. Head and Neck Pathology. 2020 Jan 4:1-9.
41. Luo W, Li S, Peng B, Ye Y, Deng X, Yao K. Embryonic stem cells markers SOX2, OCT4 and Nanog expression and their correlations with epithelial-mesenchymal transition in nasopharyngeal carcinoma. PloS one. 2013 Feb 12;8(2):e56324.
42. Keshavarz M, Asadi MH. Long non-coding RNA ES 1 controls the proliferation of breast cancer cells by regulating the Oct4/Sox2/miR-302 axis. The FEBS journal. 2019 Jul;286(13):2611-23.
43. Kaufhold S, Garbán H, Bonavida B. Yin Yang 1 is associated with cancer stem cell transcription factors (SOX2, OCT4, BMI1) and clinical implication. Journal of Experimental & Clinical Cancer Research. 2016 Dec;35(1):1-4.
44. De Bacco F, Casanova E, Medico E, Pellegatta S, Orzan F, Albano R,et al. The MET oncogene is a functional marker of a glioblastoma stem cell subtype. Cancer Research. 2012 Sep 1;72(17):4537-50.
45. Li Y, Li A, Glas M, Lal B, Ying M, Sang Y, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proceedings of the National Academy of Sciences. 2011 Jun 14;108(24):9951- 6.
46. Joo KM, Jin J, Kim E, Kim KH, Kim Y, Kang BG, et al. MET signaling regulates glioblastoma stem cells. Cancer Research. 2012 Aug 1;72(15):3828-38.
47. Rath P, Lal B, Ajala O, Li Y, Xia S, Kim J, et al. In vivo c-Met pathway inhibition depletes human glioma xenografts of tumor-propagating stem-like cells. Translational oncology. 2013 Apr 1;6(2):104-IN1.
48. Melillo G. Targeting hypoxia cell signaling for cancer therapy. Cancer and Metastasis Reviews. 2007 Jun 1;26(2):341-52.
49. Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H,et al. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proceedings of the National Academy of Sciences. 2011 Jul 26;108(30):12425- 30.
50. Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007 Jan 1;11(1):69-82.
51. Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, et al. A specialized vascular niche for adult neural stem cells. Cell Stem Cell. 2008 Sep 11;3(3):279-88.
52. Zhao M, van Straten D, Broekman ML, Préat V, Schiffelers RM. Nanocarrier-based drug combination therapy for glioblastoma. Theranostics. 2020;10(3):1355.
53. Esquela-Kerscher A, Slack FJ. Oncomirs—microRNAs with a role in cancer. Nature Reviews Cancer. 2006 Apr;6(4):259-69.
54. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nature Reviews Drug discovery. 2017 Mar;16(3):203.
55. Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature. 2010 Sep;467(7311):86-90.
56. Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019 Nov 14;179(5):1033-55.
57. Lopez-Bertoni H, Kotchetkov IS, Mihelson N, Lal B, Rui Y, Ames H, et al. A Sox2: miR-486-5p axis regulates survival of GBM cells by inhibiting tumor suppressor networks. Cancer Research. 2020 Apr 15;80(8):1644-55.
58. Lopez-Bertoni H, Kozielski KL, Rui Y, Lal B, Vaughan H, Wilson DR, et al. Bioreducible polymeric nanoparticles containing multiplexed cancer stem cell regulating miRNAs inhibit glioblastoma growth and prolong survival. Nano Letters. 2018 Jun 21;18(7):4086-94.
59. Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009 Feb 20;136(4):763-76.
60. Kim B, Park JH, Sailor MJ. Rekindling RNAi therapy: materials design requirements for in vivo siRNA delivery. Advanced Materials. 2019 Dec;31(49):1903637.
61. Lopez-Bertoni H, Kozielski KL, Rui Y, Lal B, Vaughan H, Wilson DR, et al. Bioreducible polymeric nanoparticles containing multiplexed cancer stem cell regulating miRNAs inhibit glioblastoma growth and prolong survival. Nano letters. 2018 Jun 21;18(7):4086-94.
62. Tzeng SY, Green JJ. Therapeutic nanomedicine for brain cancer. Therapeutic delivery. 2013 Jun;4(6):687-704.
63. Kozielski KL, Tzeng SY, Green JJ. Bioengineered nanoparticles for siRNA delivery. Wiley Interdisciplinary Reviews: Nanomedicine and Nanobiotechnology. 2013 Sep;5(5):449-68.
64. Lopez-Bertoni H, Kotchetkov IS, Mihelson N, Lal B, Rui Y, Ames H, et al. A Sox2: miR-486-5p axis regulates survival of GBM cells by inhibiting tumor suppressor networks. Cancer research. 2020 Apr 15;80(8):1644-55.
65. Ramos P, Bentires-Alj M. Mechanism-based cancer therapy: resistance to therapy, therapy for resistance. Oncogene. 2015 Jul;34(28):3617-26.
66. Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003 Dec 26;115(7):787-98.
67. Bartel DP. MicroRNAs: target recognition and regulatory functions. cell. 2009 Jan 23;136(2):215-33.
68. Vinogradov S, Wei X. Cancer stem cells and drug resistance: the potential of nanomedicine. Nanomedicine. 2012 Apr;7(4):597-615.
69. Vaughan HJ, Green JJ, Tzeng SY. Cancer-Targeting Nanoparticles for Combinatorial Nucleic Acid Delivery. Advanced Materials. 2020 Apr;32(13):1901081.
70. Chen KT, Wei KC, Liu HL. Theranostic strategy of focused ultrasound induced blood-brain barrier opening for CNS disease treatment. Frontiers in pharmacology. 2019 Feb 7;10:86.
71. Mitchell KA, Anderson W, Shay T, Huang J, Luciano M, Suarez JI, Manson P, Brem H, Gordon CR. First-inhuman experience with integration of wireless intracranial pressure monitoring device within a customized cranial implant. Operative Neurosurgery. 2020 Sep 1;19(3):341-50.