Abstract
Abnormal activation of epidermal growth factor receptor (EGFR) promotes the development of Non-Small Cell Lung Cancer Cells (NSCLC). Chemoresistance to tyrosine kinase inhibitors (TKIs), which is elicited by EGFR mutations, is a key challenge for NSCLC treatment. In the present study, we demonstrate a critical role of gasdermin E (GSDME), an important protein for pyroptosis, in the maintenance of EGFR stability and activation. We found that GSDME depletion suppressed the EGFR-mediated proliferation of NSCLC cells in vitro. GSDME knockdown downregulated the protein level of CCND1 and inhibited the phosphorylation of ERK1/2 in NSCLC cells. Mechanistically, both GSDME-FL and GSDME-N fragment physically interacted with EGFR. GSDME interacted with cytoplasmic fragment (CT) of EGFR. GSDME knockdown inhibited EGFR dimerization and phosphorylation at tyrosine 1173 (EGFRY1173), which could activate ERK1/2. GSDME knockdown promoted EGFR degradation and phosphorylation at tyrosine 1045 (EGFRY1045). Importantly, GSDME-FL increased the stability of EGFR, while the GSDME-N fragment induced EGFR degradation. Together, our results demonstrate that the GSDME-EGFR interaction plays an important role in NSCLC development, reveal a previously unrecognized link between GSDME and EGFR stability and offer new insight into cancer pathogenesis.
Keywords
GSDME, EGFR, ERK1/2, Proliferation
Commentary
Gasdermin E (GSDME) is specifically cleaved by caspase-3 at Asp270 with its linker, releasing the GSDME-N terminal fragment (pore-forming domain) for plasma membrane disruption and thereby inducing pyroptosis which is regarded as an inflammatory form of programmed cell death executed by the gasdermin protein family, which is characterized by cellular swelling, lysis, and release of pro-inflammatory molecules [1,2]. Several anti-cancer agents promoting the death of cancer cells through inducing pyroptosis, and previous studies of GSDME have centered on its role in pyroptosis [1-5]. In recent years, more and more research is paying attention to the role of GSDME beyond pyroptosis [6,7]. In our recent work [8], we discovered a previously unrecognized link between GSDME and EGFR (Epidermal growth factor receptor) stability and offered new insight into cancer pathogenesis.
EGFR is a receptor tyrosine kinase with fundamental roles in development and normal physiology of epithelial cells, including stimulating cell proliferation, motility and differentiation. Due to its frequent overexpression and hyperactivation, EGFR has been a therapeutic target for many epithelial cancers [9]. EGFR mutations are the second most common oncogenic driver event in NSCLC (non-small cell lung cancer) [10]. In this study, we demonstrated that GSDME physically interacts with the cytoplasmic fragment (CT) of EGFR by performing immunoprecipitation. Molecular docking analysis showed that 6 hydrogen bonding pairs (Y801EGFR-K98GSDME, Y813EGFR-T94GSDME, T993EGFR-R461GSDME, T993EGFR-K463GSDME, H998EGFR-L96GSDME and H998EGFR-K463GSDME) play a key role in the binding of GSDME to EGFR. We then performed a series of cellular assays to demonstrate that GSDME-FL (GSDME full length) acts as a positive regulator of EGFR in NSCLC cells. GSDME knockdown inhibited EGFR dimerization and phosphorylation at tyrosine 1173 (EGFRY1173), which activated ERK1/2 and promoted EGFR degradation via phosphorylation at tyrosine 1045 (EGFRY1045). Moreover, GSDME-FL increased the stability of EGFR, while the GSDME-N (GSDME-N terminal fragment) induced EGFR degradation. Overexpression of GSDME-N reduced the stability of EGFR. Therefore, different forms of GSDME might play a distinct role in tumor progression.
We observed that GSDME depletion reduced proliferation of NSCLC cells and caused G0/G1 cell cycle arrest in vitro. The protein level of CCND1 (a key G1/S transition-related protein) was notably decreased in GSDME knockdown cells. The phosphorylation of ERK1/2 (an upstream regulator of CCND1) also decreased in GSDME knockdown cells. Therefore, in addition to acting as the executioner of pyroptosis, the cytotoxic effect of GSDME depletion might be further amplified by the EGFR-ERK1/2 cascade, which affects cell survival and proliferation.
Phosphorylation of EGFRY1045 acts as the degron of EGFR and is recognized by c-Cbl (an E3 ubiquitin ligase that induces monoubiquitination and lysosome-mediated degradation of EGFR), enhancing the ubiquitination and degradation of EGFR. We speculated that GSDME-EGFR interaction covers the EGFR Y1045 site, increasing its stability and causing the abnormal activation of EGFR, while the GSDME-N fragment binds to EGFR on the membrane and promotes its degradation. The function of the GSDME-N fragment might be related to its pore-forming function, and further studies are needed to investigate the role of the GSDME-N fragment. We propose that GSDME-FL supports cell proliferation through continuously activating EGFR signaling pathway when cells are not exposed to drug treatment. However, chemotherapeutic agents’ exposure induce the cleavage of GSDME, and GSDME-N translocate to the membrane to induce pyroptosis, interacting with EGFR for degradation.
In the past decades, anticancer immunotherapies such as immune-checkpoint inhibitors (ICIs) targeting programmed cell death 1 (PD-1), programmed cell death 1 ligand 1 (PD-L1) and the inhibitory immune-checkpoint receptors cytotoxic T lymphocyte antigen 4 (CTLA-4) have demonstrated clinical efficacy in many types of cancer, including NSCLC [11,12]. However, many patients have innate or acquired resistance to immunotherapies [13]. Pyroptosis has been regarded as an immunogenic cell death which could enhance the infiltration of the CD8+ T cells in tumor tissue [14]. Therefore, it is essential to combine pyroptosis inducers with immunotherapies to increase therapeutic efficacy and improve the outcome of patients, particularly the ones with EGFR overexpression. The pyroptosis inducers may degrade EGFR to synergize the efficacy of immunotherapy. However, whether pyroptosis inducers are effective in the patients with EGFR mutations remain to be fully elucidated.
Recent study revealed that GSDME expression enhances the number and functions of tumor-infiltrating NK and CD8+ T killer and lymphocytes and tumor-associated macrophage phagocytosis. Induction of inflammatory death in GSDME-expression cancers subjected to extrinsic challenges (for example radiation, chemotherapy, and cytotoxic lymphocyte attack) or intrinsic stress (ER stress and hypoxia) that activate caspase-3 could have profound effect on tumor growth, tumor microenvironment (TME), and immune cell recruitment and function [15].
In summary, our recent work demonstrates a connection between intracellular EGFR signals and GSDME expression using a series of well-executed in vitro approaches. In addition to its role in pyroptosis, GSDME-mediated EGFR stabilization contributes to the development of NSCLC by activating the ERK1/2 pathway. GSDME-N fragment cleaved by caspase-3 perforates membranes and thereby induces pyroptosis. Targeting GSDME to release GSDME-N fragment will be benefit cancer treatment. Although some chemotherapeutic agents can induce GSDME cleavage, NSCLC often exhibits resistance to chemotherapy which may be due to the activation of other compensatory signaling pathways for cell survival. Further studies are still needed to investigate the molecular mechanism of chemotherapeutic resistance. GSDME-mediated EGFR stability may promote the proliferation of NSCLC cells, providing new insights for the future development of NSCLC therapeutic strategies to target EGFR stability via GSDME-mediated pyroptosis. Nevertheless, future studies are clearly warranted to demonstrate the regulatory mechanism of GSDME and EGFR and evaluate the feasibility of translating these promising findings into a new strategy for use in the treatment of cancer.
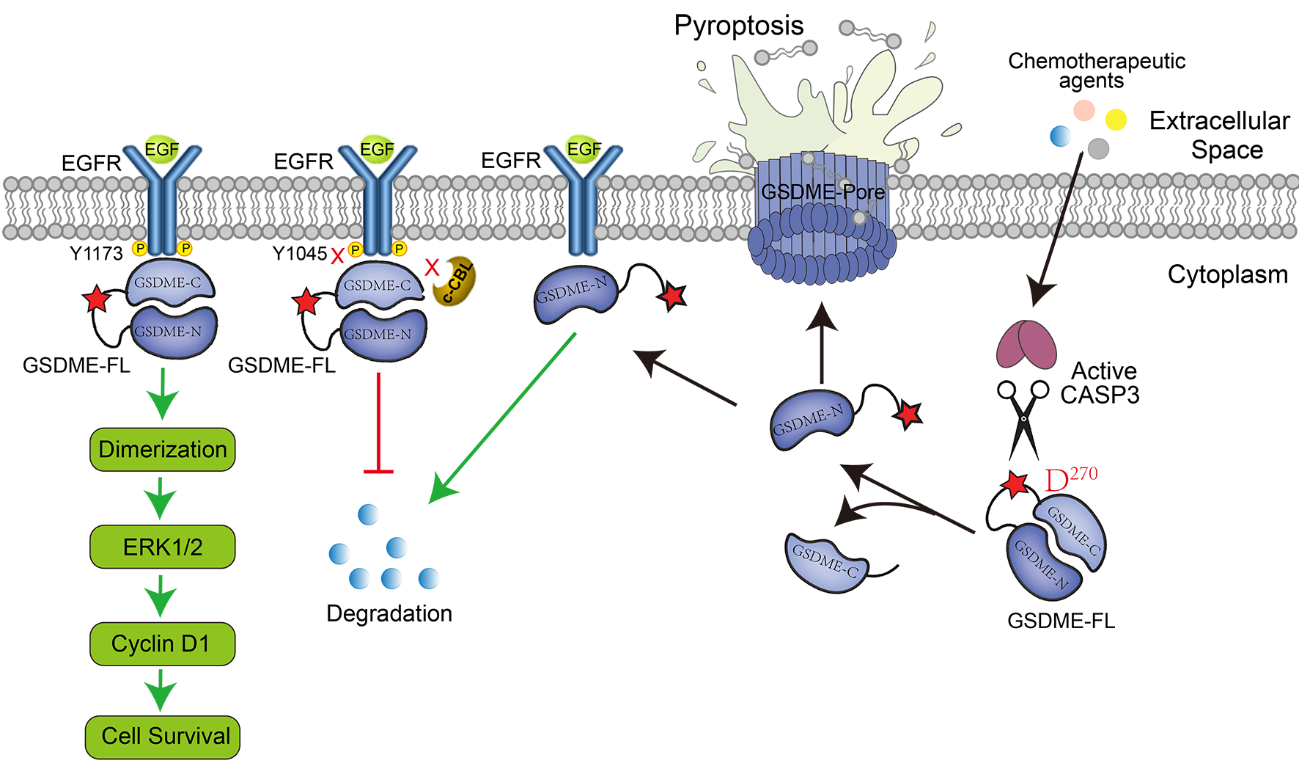
Figure 1. Working model of GSDME in regulating EGFR stability. Full-length GSDME (GSDME-FL) promotes cell proliferation by promoting EGFR dimerization and maintaining EGFR signaling. GSDME-FL maintains EGFR signaling by physically interacting with EGFR and covering the EGFRY1045 site. When cells are treated with chemotherapeutic agents, caspase-3 cleaves GSDME at Asp270 and releases the GSDME N-terminal fragment (pore-forming domain). Cleavage allows the GSDME-N to induce pyroptosis. Moreover, GSDME-N binds to EGFR on the membrane and promotes EGFR degradation.
Acknowledgements
This study was supported by the Natural Science Foundation of Shandong (ZR2022QC205), the fellowship of the China Postdoctoral Science Foundation (2021M701995), the Application Research Project of Qingdao Postdoctoral Researchers (61200071311128) and the Natural Science Foundation of China (31771526).
Competing Interests
The authors declare that they have no competing interests.
References
2. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017 Jul 6;547(7661):99-103.
3. Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Research. 2018 Dec;28(12):1171-85.
4. Han J, Cheng C, Zhang J, Fang J, Yao W, Zhu Y, et al. Myricetin activates the Caspase-3/GSDME pathway via ER stress induction of pyroptosis in lung cancer cells. Frontiers in Pharmacology. 2022 Aug 26;13:959938.
5. Fan CY, Ye FH, Peng M, Dong JJ, Chai WW, Deng WJ, et al. Endogenous HMGB1 regulates GSDME-mediated pyroptosis via ROS/ERK1/2/caspase-3/GSDME signaling in neuroblastoma. American Journal of Cancer Research. 2023;13(2):436-51.
6. Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. Journal of Hematology & Oncology. 2020 Dec;13:149.
7. Wang S, Zhang MJ, Wu ZZ, Zhu SW, Wan SC, Zhang BX, et al. GSDME is related to prognosis and response to chemotherapy in oral cancer. Journal of Dental Research. 2022 Jul;101(7):848-58.
8. Xu L, Shi F, Wu Y, Yao S, Wang Y, Jiang X, et al. Gasdermin E regulates the stability and activation of EGFR in human non-small cell lung cancer cells. Cell Communication and Signaling. 2023 Apr 21;21(1):83.
9. Tan X, Lambert PF, Rapraeger AC, Anderson RA. Stress-induced EGFR trafficking: mechanisms, functions, and therapeutic implications. Trends in Cell Biology. 2016 May 1;26(5):352-66.
10. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Seminars in Cancer Biology. 2020 Apr 1;61: 167-179.
11. Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015 Apr 3;348(6230):56-61.
12. Zappasodi R, Merghoub T, Wolchok JD. Emerging concepts for immune checkpoint blockade-based combination therapies. Cancer Cell. 2018 Apr 9;33(4):581-98.
13. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treatment Reviews. 2017 Jan 1;52:71-81.
14. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature. 2020 Mar 19;579(7799):421-6.
15. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020 Mar 19;579(7799):415-20.