Abstract
TLR4, as an on-membrane receptor of LPS, plays a crucial part in the process of sepsis, and EGFR phosphorylation promotes cell inflammation in response to LPS, which plays an indispensable role in modulating LPS/TLR4 signaling pathway. However, the mechanism of interaction between TLR4 and EGFR signaling is still unclear. In addition, there is an inextricable relationship between the activation of EGFR and the signaling pathway where Rab is located. This review mainly summarizes the research progress of the relationship among TLR4, EGFR and members of the Rab family, especially Rab5a under LPS stimulation, which also involves other signaling pathways of EGFR and Rab proteins.
Keywords
LPS, Sepsis, TLR4, EGFR, Rab family
Introduction
Lipopolysaccharide (LPS), an abundant molecule of the outer membrane of most but not all Gram-negative bacteria, plays a vital role during host–pathogen interaction and the pathogenesis of infection [1]. Therefore, LPS is commonly stimulated in basic experiments to construct in vivo and in vitro sepsis models for exploring the internal molecular mechanism of sepsis.
Toll-like receptor 4 (TLR4), which is the most investigated member of the Toll-like receptor family, is a crucial receptor sensing bacterial LPS [2]. In bacterial infection, cell surface expression of TLR4 on innate immune cells critically regulates host responses and inflammatory processes [3,4], moreover, its dysregulation is thought to promote aberrant cytokine production in bacterial sepsis [5].
Epidermal growth factor receptor (EGFR) is a member of the tyrosine kinase receptor family, which is composed of extracellular, transmembrane, and intracellular domains [6]. After binding to its ligands, including epidermal growth factor (EGF), transforming growth factor α (TGF-α) and amphiregulin (AR), EGFR plays an essential role in regulation of cell proliferation, differentiation and motility [7]. We and the other researchers have previously reported that EGFR can be transactivated by LPS, which is required for LPS-induced NF-κB activation, in addition that EGFR phosphorylation promotes LPS-induced TNF-α production [8-10], and next comes cell inflammatory response, while the EGFR reversible inhibitor erlotinib effectively prevents LPS-induced cytokine expression in vivo with likewise efficaciously protecting mice from LPSinduced lethality [9,11,12]. These findings suggest the cross-talk between TLR4 and EGFR signaling pathways, which importantly affects the inflammatory progression and host prognosis following bacterial infection.
Thus, it follows that understanding the TLR4/EGFRrelated signaling pathways will provide essential information about the basic mechanisms underlying inflammation and sepsis. In this article, we review the literature on the mechanism underlying the cross-talk between TLR4 and EGFR signaling, with special emphasis on summarizing the research progression of EGFR and Rab protein family signaling pathways.
Interconnection between TLR4 and EGFR
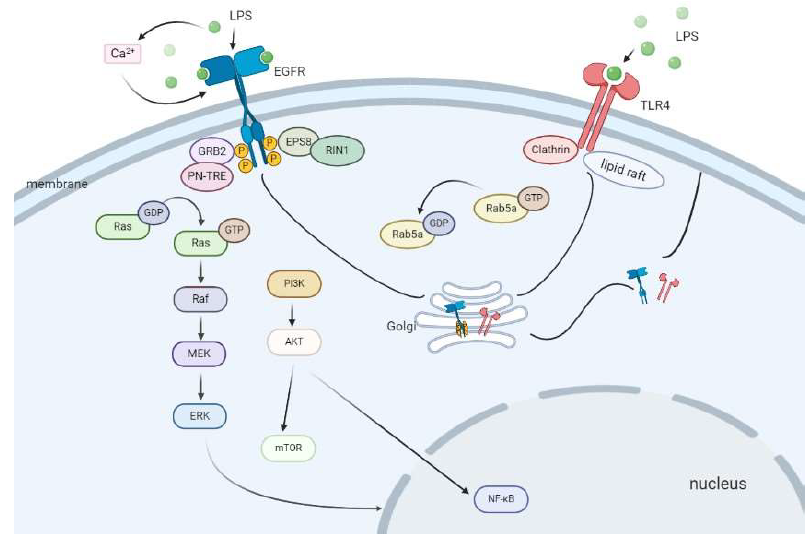
The crucial link between TLR4 and EGFR is described above, what’s more interesting is that our current finding reveals that EGFR-dependent initiation of internalization of TLR4 and EGFR activates a cascade of intracellular events leading to enhanced cell surface expression of TLR4 rather than desensitization of the cells to LPS, which represents a shift in our understanding of the significance of receptor endocytosis. And in this study, we demonstrate, for the first time, that LPS also increases the EGFR expression on the cell surface of macrophage [13]. This may explain, at least partially, why LPS can also increase EGFR-mediated effects such as cell proliferation [14,15]. Most previous studies focus on the process of receptor endocytosis or the trafficking of receptors from the Golgi apparatus to the cell surface [16,17]. Our nearterm study provides evidence that LPS-induced early endocytosis of TLR4 is critical for later increased cell surface expression of TLR4. In fact, shuttling of TLR4 from the cell surface to the Golgi and back to the cell surface occurs dynamically after LPS stimulation. These two processes are tightly connected and mutually interact with each other. As we block endocytosis of LPS/TLR4 complex with clathrin inhibitor chlorpromazine (CPZ) or EGFR inhibitor PD168393, the increase of TLR4 on the cell surface in response to LPS is significantly decreased [13]. However, we still do not fully understand the detailed mechanism underlying how endocytosis of TLR4 increases transportation of TLR4 from the Golgi apparatus onto the cell membrane. CPZ or PD168393 pretreatment does not completely block the increase of TLR4 on the cell surface in response to LPS. This suggests that some other signaling pathways may also be involved in the regulation of TLR4 surface expression besides clathrin and EGFR. For example, one study has shown that LPS induces TLR4 internalization through clathrin-independent caveolae/ lipid raft-mediated pathways [18]. A study by Husebye et al. suggests that in the early phases after LPS stimulation, TLR4 internalization is predominantly clathrin-dependent, whereas clathrin-independent pathways operate at later time points [19]. Our belief is that TLR4 endocytosis and trafficking onto cell surface occur simultaneously in response to LPS stimulation, and the net amount of the cell surface expression depends on the dynamic balance of the two processes. In addition, LPS stimulation may not play an inflammatory role through the TLR4-EGFR pathway under a specified condition but may induce EGFR phosphorylation by other means to initiate downstream inflammatory pathways. For example, LPS could trigger the rapid phosphorylation of EGFR and subsequent ERK activation through mobilizing calcium activity, which underlies the microglia migration in an inflammatory condition. Otherwise, inhibition of calcium oscillation suppresses EGFR phosphorylation in microglia, which as a result, depresses LPS-induced migration of microglia [20].
In addition to sepsis, TLR4/EGFR also participates in the pathophysiological processes of tumors, cardiovascular system diseases and other inflammations, and all of them, to a certain extent, exist not a few common points in molecular mechanisms, which probably provides us quite a number of experimental ideas in research and clinical treatments. According to Houyu Ju et al, EGFR is highly expressed in head and neck squamous cell carcinoma (HNSCC) and relates to cancer progression, while TLR4 is highly expressed in 50% of EGFR overexpressed HNSCC biopsies, which correlates to worse prognosis in patients. In HNSCC cell lines, activation of TLR4 reverses cetuximab-induced inhibition of proliferation, migration, and invasion. Mechanistically, inhibition of EGFR by cetuximab leads to decreased phosphorylation of Src and sequentially Src-medicated activation of Cbl-b, which inhibits Cbl-b-mediated degradation of the key TLR4 adaptor protein MyD88 and activates TLR4 signaling. The study delineates a crosstalk between EGFR and TLR4 pathways and identifies TLR4 that acts as a potential biomarker as well as a therapeutic target in overcoming the resistance to anti-EGFR therapy of HNSCC [21]. The highly concerned COVID-19 infection, which is very difficult to distinguish between from EGFR tyrosine kinase inhibitor (TKI)–associated interstitial lung disease (ILD) with clinical and imaging presentations [22], may develop into sepsis, when in the case of poor control. Consequently, non-specific targeted drugs targeting EGFR for the treatment of related pneumonia, taking vitamin C as an example, may have potential markets [23]. Meanwhile, these research results above remind us whether COVID-19 pathogenesis is inextricably linked to the TLR4/EGFR pathway?
The Role of Rab5a in LPS-induced EGFR Activation
About 70 members of Rab proteins are encoded in human genome. Rab proteins constitute the largest family in the Ras small GTPase superfamily, which regulates different stages of intracellular membrane trafficking including vesicle budding, transportation, docking and fusion [24-26]. Members of the Rab GTPases are localized to various intracellular compartments, where they control vesicular trafficking through their interactions with specific tethering proteins and the cytoskeleton. Mechanistically, Rab protein cycles between GTP-bound active form and GDP-bound inactive forms. Videlicet, activation of Rab protein is facilitated by Guanine nucleotide exchange factors (GEFs) which stimulate GDP exchange for GTP, whereas inactivation of Rab protein is stimulated by GTPase activating proteins (GAPs) which promote GTP hydrolysis. GTP-bound active Rab protein interacts with their downstream effectors to mediate multiple biological functions [27].
Among the Rab GTPases, Rab5 which localizes to early endosomes, the plasma membrane, and clathrin-coated vesicles is involved in the control of intracellular trafficking, largely between the plasma membrane and the endosomal compartment [28]. Rab5 is one of the best characterized members and one of the endosomal Rab proteins [29]. In addition to its role in endocytosis, Rab5 has been implicated in other cellular processes, such as cell adhesion and migration, cell mitosis, autophagy, and so on [30,31]. Rab5 includes three isoforms, Rab5a, Rab5b, and Rab5c respectively. Rab5a localizes on both plasma membrane and endocytic vesicles [32]. In its GTP-bound state, Rab5a is membrane-associated, whereas GDP-bound Rab5a is found in a cytosolic complex with the general Rab effector Rab guanine nucleotide dissociation inhibitor (GDI) [33]. As an important isoform of Rab5, Rab5a plays a crucial role in the internalization of plasma membrane proteins and their return to the cell surface. It not only regulates G protein-coupled receptor (GPCR) trafficking, but also mediates junction protein localization [34,35].
Rab5a seems to be a key molecule mediating the circulation of TLR4 and EGFR from the cell surface and back in response to LPS. LPS induces EGFR autophosphorylation, creating docking sites for the recruitment of its substrate EPS8 and SH2 domain-containing signaling molecules, such as Grb2 [36,37]. This is followed by activation of Ras effector RIN1 and Rab5 GTPase-activating protein (GAP) RN-TRE [38]. RIN1 encodes a guanine nucleotide exchange factor (GEF) domain which activates Rab5 GTPases inducing membrane ruffling and macropinocytosis, elevating GTPRab5 levels and governing early endosome traffic [39,40]. RN-TRE is a binding partner for the EGFR substrate Eps8 and contains a Rab family GAP homology domain for Rab5. There is a direct interaction between RN-TRE and GTP-bound Rab5 [36,37]. In its GTP-bound form, Rab5 recruits cytosolic factors, such as EEA1 and Rabaptin-5, to promote endosome docking and fusion. On the other hand, recycling of Rab5a to the GDP-bound state is essential for normal trafficking [37]. EGFR phosphorylation activates both RIN1 and RN-TRE which work together to maintain the equilibrium and accelerate the circulation of GTPbound and GDP-bound Rab5a. In this way, Rab5a can work constantly to transport TLR4 and EGFR from the cell surface into early endosomes. When we inhibit the phosphorylation of EGFR, the activation of Rab5a is inhibited, which leads to the dysfunction of both TLR4 and EGFR endocytosis. EGFR phosphorylation regulates the activity of Rab5a. As shown in our research, either EGFR inhibitor or knockdown of EPS8, GRB2, RIN1, RN-TRE and Rab5a expression effectively decreases the expression of both EGFR and TLR4 on the cell surface induced by LPS. The amount of TLR4 on the cell surface can to some extent determine the intensity of the inflammatory response induced in cells stimulated by LPS. This may explain why EGFR inhibitor can suppress the activation of p38 and ERK1/2, the expression of inflammatory cytokines, and the production of ROS in response to LPS [13].
Character of Other Rab Members in EGFR-related Signaling Pathways
Rab7 is a major regulator of a variety of intracellular trafficking steps that includes endosomal maturation, transporting from the late endosome to the lysosome, endolysosomal positioning via regulation of trafficking of cargo along microtubules, retromer-mediated trafficking, lysosome reformation and autophagosome maturation [41,42].
Patricia Gómez-Suaga et al. describe that leucine-rich repeat kinase 2 (LRRK2) delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity with a concomitant deficit in Rab7-mediated membrane trafficking events, which leads to a delay in the trafficking of the EGFR out of late endosomes that become aberrantly elongated, and a delay in degradation of the EGFR [41]. Interestingly, the effects of pathogenic LRRK2 can be rescued by overexpressing wild-type CIN85 (cbl-interacting protein of 85 kDa) and dynamin 2 (dyn2), while inhibiting the interaction of CIN85–dyn2 prevents exit of the EGFR out of late endosomes [43], similarly to what is observed when expressing a Rab7 dominant-negative mutant or knocking down Rab7 [44]. Leucine-rich repeat kinase1 (LRRK1) phosphorylates the conserved S72 residue within the switch II region of Rab7 (as a substrate of LRRK1), which increases the association of Rab7 with its effector RILP in turn and results in enhanced minus-end transport of EGFR-containing endosomes through a dynein-dependent mechanism [45,46], while the hyperactivation of LRRK1 kinase stimulates the motility of EGFR-containing endosomes toward the nucleus and causes the immature perinuclear clustering of EGFR-containing endosomes, which cannot fuse with lysosomes [47].
Tumour suppressor phosphatase and tensin homologue (PTEN), as a core regulator of PI3-kinase signalling network, controls endocytic trafficking of EGFR by promoting late endosome maturation, which means that PTEN is required for efficient transition of ligand-bound EGFR from early to late endosomes. Complementarily, PTEN dephosphorylates Rab7 and regulates its localization to the late endosomal membranes, which is critical for endosome maturation [48].
Wang et al. find that CD44s splice isoform attenuates endocytosis-mediated EGFR degradation, thus sustaining downstream Akt signaling, while Rab7a acts as a major downstream target for CD44s in mediating EGFR degradation [49].
Rab7, as a substrate of Src kinase, is tyrosine phsphorylated at Y183 residue, which is physiologically regulated by EGF, inhibits the degradation of EGFR and sustains AKT signaling [25], meanwhile, Rab7 is supposed to sustain Akt survival signal by preventing Hsp90 dependent EGFR degradation [50].
Rab7b, highly similar to Rab7, is involved in the regulation of transport to degradative compartments in the endocytic pathway [51]. Interestingly, expression of a Rab7b dominant-negative mutant or silencing of Rab7b in HeLa cells does not alter EGF or EGFR degradation, while Rab7b promotes degradation of the receptor TLR4, unlike EGFR, is not only destined for the degradative multivesicular late endosomes but also to other compartments, such as the Golgi apparatus, from where it can recycle to the plasma membrane [19]. Therefore, Rab7b could impair TLR4 cycling between the Golgi and endosomes, which may affect the degradation and regulate the function of this receptor [52].
Amongst Rab GTPases, Rab1, the mammalian homologue of yeast protein transport 1 (Ypt1), is a central regulator of dynamic membrane trafficking between endoplasmic reticulum (ER) and Golgi apparatus in the secretory pathway. Beyond their classical vesicular transport functions, Rab1 proteins also regulate expression of cellsurface receptors, cell migration, and nutrient sensing [53]. There are two Rab1 isoforms, to wit, Rab1a and Rab1b which share 92% amino-acid sequence homology with most differences in the carboxyl terminus. Rab1a and Rab1b are mainly expressed at the ER and the Golgi membrane, and are also detected in lipid rafts [54] and autophagosomes [55]. A recent work provides strong evidence that Rab1a activation is crucial for the production of proinflammatory cytokines IL-1β and IL-18 in macrophages and subsequent lung inflammatory injury during sepsis. Rab1a regulates the release of IL-1β and IL-18 via priming macrophages by up-regulating pro-IL-1β and pro-IL-18 expression and stimulating TLR4/NF-κB signaling by promoting TLR4 trafficking from ER to cell membrane and activates [56].
It is reported that in macrophages, Rab20, associated with early endocytic organelles, mainly localizes in the Golgi complex and early endosomes, and its expression induces the formation of enlarged endosomes. Of importance whereas both endocytic uptake of transferrin and macropinocytosis are not affected by the expression of the wild-type and dominant-negative forms of Rab20, Rab20 knockdown accelerates EGF–stimulated endocytic trafficking to LAMP-2–positive compartments and degradation of EGFR [57].
Furthermore, Ras and Rab interactor 1 (RIN1) enhances clear cell renal cell carcinoma (ccRCC) cell growth, migration and invasion abilities in vitro and promotes tumor growth and metastasis in vivo by activation of EGFR signaling through interacting with Rab25 [58]. Strikingly, Jeong et al. demonstrate that Rab25 increases β1 integrin levels and subsequent activation of EGFR and upregulation of vascular endothelial growth factor (VEGF)-A expression, leading to increased Snail expression, epithelial-tomesenchymal transition and cancer cell invasiveness [59].
Hsieh et al.’s study suggests that the expression of RAB38 may be associated with EGFR status since RAb38 is upregulated in cases of NSCLC associated with an active EGFR mutation, compared with those associated with wild-type EGFR [60].
Overexpression of Rab11-FIP2 (a member of a family of Rab11-binding proteins) may result in suppression of the internalization of EGFR in COS-7 cells [61] as well as promoting PI3K/AKT activation and migratory capacities of AGS/FIP2 cells induced by EGF in gastric cancer, which can be inhibited by LY294002 (an inhibitor of PI3K/AKT) [62].
It is reported that Rab21 enhances the degradation of EGFR through accelerating its internalization in both EGF-independent and EGF-dependent manners, resulting in attenuating EGF-mediated mitogen-activated protein kinase (MAPK) signaling [63].
Chua et al. suggest that Rab31 inhibits, but overexpression enhances, EGFR trafficking to the late endosomes, likewise, the effect of Rab31 silencing can be specifically rescued by overexpression of a silencing-resistant form of Rab31. And interestingly, the loss of early endosome antigen 1 (EEA1) or GAPex5 (a Rab31 guanine nucleotide exchange factor) reduces the interaction of Rab31 with the EGFR and its effect on EGFR trafficking [64].
A recent study demonstrates that the extensive characterization of early endosomes in breast cancer cells identifies a Rab4-modulated enlarged early endosomal compartment as the site of prolonged and increased EGFR activation by comparing MDA-MB-231 breast cancer cells with noncancerous MCF10A cells [65].
Conclusions
Taken together, the expression of TLR4 and EGFR increases on the cell membrane after LPS stimulation. Although there is no direct correlation between TLR4 and EGFR, both may use the same intracellular transport mechanism, which results that the exact mechanism of the interaction between EGFR and TLR4 remains to be further explored. Rab proteins, involved in the regulation of membrane trafficking, is inseparable from intracellular trafficking and degradation of EFGR. And our own study elucidates a novel mechanism that Rab5a plays an important role in promoting macrophage surface expression of TLR4 and EGFR after LPS stimulation. In addition to Rab5a, various other members of Rab family, such as Rab7a, Rab8a, Rab1a, etc, are also involved in the signaling pathways and cascades initiated by EGFR in inflammation. Over and above that, there are plenty of studies on tumors regarding the relationship between EGFR and the Rab family, while existing similarities in the occurrence and development between tumors and inflammation, which suggests whether we can refer from these results to excavate new mechanisms of Rab proteins and EGFR in inflammation, thereby carrying on in-depth exploration of the mechanism and clinical targeted therapy of sepsis.
Funding Statements
This work was supported by the National Natural Science Foundation of China 81671957 and 81873951 (J.T.), key projects of Guangdong Natural Science Foundation 2018B030311038 (J.T.), and Science and Technology Planning Project of Guangdong Province 2016A020215212 (J.T.).
Conflicts of Interest
There is no conflict of interest.
References
2. Klein DCG, Skjesol A, Kers-Rebel ED, Sherstova T, Sporsheim B, Egeberg KW, et al. CD14, TLR4 and TRAM Show Different Trafficking Dynamics During LPS Stimulation. Traffic. 2015;16(7):677-90.
3. Straub T, Freudenberg MA, Schleicher U, Bogdan C, Gasteiger G, Pircher H. Bacterial coinfection restrains antiviral CD8 T-cell response via LPS-induced inhibitory NK cells. Nat Commun. 2018;9(1):4117.
4. Parnas O, Jovanovic M, Eisenhaure TM, Herbst RH, Dixit A, Ye CJ, et al. A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell. 2015;162(3):675-86.
5. Rosadini CV, Kagan JC. Early innate immune responses to bacterial LPS. Curr Opin Immunol. 2017;44:14-9.
6. Shen M, Jiang Y-Z, Wei Y, Ell B, Sheng X, Esposito M, et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell. 2019;35(1).
7. Ye Q-H, Zhu W-W, Zhang J-B, Qin Y, Lu M, Lin G-L, et al. GOLM1 Modulates EGFR/RTK Cell-Surface Recycling to Drive Hepatocellular Carcinoma Metastasis. Cancer Cell. 2016;30(3):444-58.
8. Chattopadhyay S, Veleeparambil M, Poddar D, Abdulkhalek S, Bandyopadhyay SK, Fensterl V, et al. EGFR kinase activity is required for TLR4 signaling and the septic shock response. EMBO Rep. 2015;16(11):1535- 47.
9. De S, Zhou H, DeSantis D, Croniger CM, Li X, Stark GR. Erlotinib protects against LPS-induced endotoxicity because TLR4 needs EGFR to signal. Proc Natl Acad Sci USA. 2015;112(31):9680-5.
10. Chen W, Zhong H, Wang X, Pang Q, Zhuang J, Hu J, et al. Mig6 reduces inflammatory mediators production by regulating the activation of EGFR in LPS-induced endotoxemia. J Cell Physiol. 2018;233(9):6975-83.
11. Singer M, Deutschman CS, Seymour CW, Shankar- Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):801-10.
12. Sun X, Liang J, Yao X, Lu C, Zhong T, Hong X, et al. The activation of EGFR promotes myocardial tumor necrosis factor-α production and cardiac failure in endotoxemia. Oncotarget. 2015;6(34):35478-95.
13. Tang J, Zhou B, Scott MJ, Chen L, Lai D, Fan EK, et al. Correction to: EGFR signaling augments TLR4 cell surface expression and function in macrophages via regulation of Rab5a activation. Protein Cell. 2020.
14. Español AJ, Maddaleno MO, Lombardi MG, Cella M, Martínez Pulido P, Sales ME. Treatment with LPS plus INF-γ induces the expression and function of muscarinic acetylcholine receptors, modulating NIH3T3 cell proliferation: participation of NOS and COX. Br J Pharmacol. 2014;171(22):5154-67.
15. Takizawa H, Fritsch K, Kovtonyuk LV, Saito Y, Yakkala C, Jacobs K, et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell. 2017;21(2).
16. Watanabe S, Boucrot E. Fast and ultrafast endocytosis. Curr Opin Cell Biol. 2017;47:64-71.
17. Xu X, Xu J, Wu J, Hu Y, Han Y, Gu Y, et al. Phosphorylation-Mediated IFN-γR2 Membrane Translocation Is Required to Activate Macrophage Innate Response. Cell. 2018;175(5).
18. Triantafilou M, Morath S, Mackie A, Hartung T, Triantafilou K. Lateral diffusion of Toll-like receptors reveals that they are transiently confined within lipid rafts on the plasma membrane. J Cell Sci. 2004;117(Pt 17):4007-14.
19. Husebye H, Halaas Ø, Stenmark H, Tunheim G, Sandanger Ø, Bogen B, et al. Endocytic pathways regulate Toll-like receptor 4 signaling and link innate and adaptive immunity. EMBO J. 2006;25(4):683-92.
20. Qu W-S, Liu J-L, Li C-Y, Li X, Xie M-J, Wang W, et al. Rapidly activated epidermal growth factor receptor mediates lipopolysaccharide-triggered migration of microglia. Neurochem Int. 2015;90:85-92.
21. Ju H, Hu Z, Lu Y, Wu Y, Zhang L, Wei D, et al. TLR4 activation leads to anti-EGFR therapy resistance in head and neck squamous cell carcinoma. Am J Cancer Res. 2020;10(2):454-72.
22. Chang H-L, Chen Y-H, Taiwan H-C, Yang C-J. EGFR Tyrosine Kinase Inhibitor-Associated Interstitial Lung Disease During the Coronavirus Disease 2019 Pandemic. J Thorac Oncol. 2020.
23. Li R, Guo C, Li Y, Qin Z, Huang W. Therapeutic targets and signaling mechanisms of vitamin C activity against sepsis: a bioinformatics study. Brief Bioinformatics. 2020.
24. Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513-25.
25. Lin X, Zhang J, Chen L, Chen Y, Xu X, Hong W, et al. Tyrosine phosphorylation of Rab7 by Src kinase. Cell Signal. 2017;35:84-94.
26. Rojas AM, Fuentes G, Rausell A, Valencia A. The Ras protein superfamily: evolutionary tree and role of conserved amino acids. J Cell Biol. 2012;196(2):189-201.
27. Goody RS, Rak A, Alexandrov K. The structural and mechanistic basis for recycling of Rab proteins between membrane compartments. Cell Mol Life Sci. 2005;62(15):1657-70.
28. Yang J, Yao W, Qian G, Wei Z, Wu G, Wang G. Rab5- mediated VE-cadherin internalization regulates the barrier function of the lung microvascular endothelium. Cell Mol Life Sci. 2015;72(24):4849-66.
29. Jongsma MLM, Berlin I, Wijdeven RHM, Janssen L, Janssen GMC, Garstka MA, et al. An ER-Associated Pathway Defines Endosomal Architecture for Controlled Cargo Transport. Cell. 2016;166(1):152-66.
30. Mendoza P, Ortiz R, Díaz J, Quest AFG, Leyton L, Stupack D, et al. Rab5 activation promotes focal adhesion disassembly, migration and invasiveness in tumor cells. J Cell Sci. 2013;126(Pt 17):3835-47.
31. Li Y, Zhao Y, Hu J, Xiao J, Qu L, Wang Z, et al. A novel ER-localized transmembrane protein, EMC6, interacts with RAB5A and regulates cell autophagy. Autophagy. 2013;9(2):150-63.
32. Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91(1):119-49.
33. Woodman PG. Biogenesis of the sorting endosome: the role of Rab5. Traffic. 2000;1(9):695-701.
34. Stamatovic SM, Sladojevic N, Keep RF, Andjelkovic AV. Relocalization of junctional adhesion molecule A during inflammatory stimulation of brain endothelial cells. Mol Cell Biol. 2012;32(17):3414-27.
35. Asaka M, Hirase T, Hashimoto-Komatsu A, Node K. Rab5a-mediated localization of claudin-1 is regulated by proteasomes in endothelial cells. Am J Physiol, Cell Physiol. 2011;300(1):C87-C96.
36. Lanzetti L, Rybin V, Malabarba MG, Christoforidis S, Scita G, Zerial M, et al. The Eps8 protein coordinates EGF receptor signalling through Rac and trafficking through Rab5. Nature. 2000;408(6810):374-7.
37. Martinu L, Santiago-Walker A, Qi H, Chou MM. Endocytosis of epidermal growth factor receptor regulated by Grb2-mediated recruitment of the Rab5 GTPase-activating protein RN-tre. J Biol Chem. 2002;277(52):50996-1002.
38. Balaji K, French CT, Miller JF, Colicelli J. The RAB5- GEF Function of RIN1 Regulates Multiple Steps During Listeria monocytogenes Infection. Traffic. 2015;16(7):796.
39. Barbieri MA, Fernandez-Pol S, Hunker C, Horazdovsky BH, Stahl PD. Role of rab5 in EGF receptor-mediated signal transduction. Eur J Cell Biol. 2004;83(6):305-14.
40. Balaji K, Mooser C, Janson CM, Bliss JM, Hojjat H, Colicelli J. RIN1 orchestrates the activation of RAB5 GTPases and ABL tyrosine kinases to determine the fate of EGFR. J Cell Sci. 2012;125(Pt 23):5887-96.
41. Gómez-Suaga P, Rivero-Ríos P, Fdez E, Blanca Ramírez M, Ferrer I, Aiastui A, et al. LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet. 2014;23(25):6779-96.
42. Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942-6.
43. Schroeder B, Weller SG, Chen J, Billadeau D, McNiven MA. A Dyn2-CIN85 complex mediates degradative traffic of the EGFR by regulation of late endosomal budding. EMBO J. 2010;29(18):3039-53.
44. Vanlandingham PA, Ceresa BP. Rab7 regulates late endocytic trafficking downstream of multivesicular body biogenesis and cargo sequestration. J Biol Chem. 2009;284(18):12110-24.
45. Hanafusa H, Yagi T, Ikeda H, Hisamoto N, Nishioka T, Kaibuchi K, et al. LRRK1 phosphorylation of Rab7 at S72 links trafficking of EGFR-containing endosomes to its effector RILP. J Cell Sci. 2019;132(11).
46. Kedashiro S, Pastuhov SI, Nishioka T, Watanabe T, Kaibuchi K, Matsumoto K, et al. LRRK1-phosphorylated CLIP-170 regulates EGFR trafficking by recruiting p150Glued to microtubule plus ends. J Cell Sci. 2015;128(2):385-96.
47. Ishikawa K, Nara A, Matsumoto K, Hanafusa H. EGFR-dependent phosphorylation of leucine-rich repeat kinase LRRK1 is important for proper endosomal trafficking of EGFR. Mol Biol Cell. 2012;23(7):1294-306.
48. Shinde SR, Maddika S. PTEN modulates EGFR late endocytic trafficking and degradation by dephosphorylating Rab7. Nat Commun. 2016;7:10689.
49. Wang W, Zhang H, Liu S, Kim CK, Xu Y, Hurley LA, et al. Internalized CD44s splice isoform attenuates EGFR degradation by targeting Rab7A. Proc Natl Acad Sci USA. 2017;114(31):8366-71.
50. Wang T, Zhang M, Ma Z, Guo K, Tergaonkar V, Zeng Q, et al. A role of Rab7 in stabilizing EGFR-Her2 and in sustaining Akt survival signal. J Cell Physiol. 2012;227(6):2788-97.
51. Wang Y, Chen T, Han C, He D, Liu H, An H, et al. Lysosome-associated small Rab GTPase Rab7b negatively regulates TLR4 signaling in macrophages by promoting lysosomal degradation of TLR4. Blood. 2007;110(3):962- 71.
52. Progida C, Cogli L, Piro F, De Luca A, Bakke O, Bucci C. Rab7b controls trafficking from endosomes to the TGN. J Cell Sci. 2010;123(Pt 9):1480-91.
53. Yang XZ, Li XX, Zhang YJ, Rodriguez-Rodriguez L, Xiang MQ, Wang HY, et al. Rab1 in cell signaling, cancer and other diseases. Oncogene. 2016;35(44):5699-704.
54. Wang C, Yoo Y, Fan H, Kim E, Guan K-L, Guan J-L. Regulation of Integrin β 1 recycling to lipid rafts by Rab1a to promote cell migration. J Biol Chem. 2010;285(38):29398- 405.
55. Zoppino FCM, Militello RD, Slavin I, Alvarez C, Colombo MI. Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic. 2010;11(9):1246-61.
56. Zhang Y, Wang L, Lv Y, Jiang C, Wu G, Dull RO, et al. The GTPase Rab1 Is Required for NLRP3 Inflammasome Activation and Inflammatory Lung Injury. J Immunol. 2019;202(1):194-206.
57. Pei G, Schnettger L, Bronietzki M, Repnik U, Griffiths G, Gutierrez MG. Interferon-γ-inducible Rab20 regulates endosomal morphology and EGFR degradation in macrophages. Mol Biol Cell. 2015;26(17):3061-70.
58. Feng Z-H, Fang Y, Zhao L-Y, Lu J, Wang Y-Q, Chen Z-H, et al. RIN1 promotes renal cell carcinoma malignancy by activating EGFR signaling through Rab25. Cancer Sci. 2017;108(8):1620-7.
59. Jeong BY, Cho KH, Jeong KJ, Park Y-Y, Kim JM, Rha SY, et al. Rab25 augments cancer cell invasiveness through a β1 integrin/EGFR/VEGF-A/Snail signaling axis and expression of fascin. Exp Mol Med. 2018;50(1):e435.
60. Hsieh J-J, Hou M-M, Chang JW-C, Shen Y-C, Cheng H-Y, Hsu T. is a potential prognostic factor for tumor recurrence in non-small cell lung cancer. Oncol Lett. 2019;18(3):2598-604.
61. Parachoniak CA, Luo Y, Abella JV, Keen JH, Park M. GGA3 functions as a switch to promote Met receptor recycling, essential for sustained ERK and cell migration. Dev Cell. 2011;20(6):751-63.
62. Dong W, Qin G, Shen R. Rab11-FIP2 promotes the metastasis of gastric cancer cells. Int J Cancer. 2016;138(7):1680-8.
63. Yang X, Zhang Y, Li S, Liu C, Jin Z, Wang Y, et al. Rab21 attenuates EGF-mediated MAPK signaling through enhancing EGFR internalization and degradation. Biochem Biophys Res Commun. 2012;421(4):651-7.
64. Chua CEL, Tang BL. Engagement of the small GTPase Rab31 protein and its effector, early endosome antigen 1, is important for trafficking of the ligand-bound epidermal growth factor receptor from the early to the late endosome. J Biol Chem. 2014;289(18):12375-89.
65. Tubbesing K, Ward J, Abini-Agbomson R, Malhotra A, Rudkouskaya A, Warren J, et al. Complex Rab4-Mediated Regulation of Endosomal Size and EGFR Activation. Mol Cancer Res. 2020;18(5):757-73.