Keywords
MYC, VX-680, AURKB, Bcl-2, Bcl-xL, Autophagy, Apoptosis
Targeting the MYC Oncogene
The transcription factor encoded by the myelocytomatosis oncogene (MYC) is deregulated by distinct means in different human cancers. Aberrations include chromosomal translocation, amplification, mutation, enhancer activation and post-translational mechanisms that lead to MYC protein accumulation [1]. It is perhaps the most commonly deregulated oncoprotein and is linked to most of the hallmarks of cancer [2]. Yet, despite decades of research, targeting MYC’s transforming function is still somewhat enigmatic. Without a kinase domain, it has proven difficult to target with small molecule inhibitors and since MYC is an intracellular protein, it is currently inaccessible to large molecules such as antibodies. A promising alternative approach, that has yet to be fully realized clinically, is to target MYC overexpressing cells with compounds that are synthetic lethal to MYC. Cancers that may benefit from successful synthetic lethal targeting of MYC include various leukemias and lymphomas where MYC deregulation plays a significant role in malignancy [3-5].
MYC Synthetic Lethality with AURKB Inhibition
Synthetic lethal interactions arise when specific oncogenic events impart overdependence on the integrity of specific cellular pathways and functions. Interference with these dependencies can result in dramatic anti-tumor effects. Examples of synthetic lethal interactions with MYC include cell death induced by activation of the TNF receptor superfamily member DR5 (encoded by TNFRSF10B) [6], dependence on the non-essential amino acid glutamine [7], pharmacological inhibition of CDK1 [8], PIM1 [9] or CHK1/2 kinases [10,11] and genetic depletion of AMPKrelated kinase 5 [12] or the core spliceosome component, BUD31 [13]. However, the most well studied synthetic lethality with MYC may be the enhanced sensitivity of MYC overexpressing cells to Aurora kinase B (AURKB) inhibition [14-16]. AURKB, along with the proteins INCENP, Borealin and Survivin, form the chromosomal passenger protein complex (CPPC). The kinase activity of AURKB, the catalytic subunit of the complex, is spatially and temporally regulated during mitosis through association with the other components and this is guided by the combined activities of multiple other kinases [17]. A functioning CPPC is required for key events in mitosis, including safeguarding proper chromosome-microtubule attachment, spindle assembly checkpoint activation and cytokinesis. Accordingly, transient inhibition of AURKB can induce both mitotic arrest with apoptosis in MYC expressing cells alongside the induction of polyploidy from failed cytokinesis in cells that escape the earlier fate [14].
Recently, MYC has been linked to enhanced expression of genes impacting centrosome function and chromosomal instability (CIN) arising from errors in chromosome segregation [18]. Key proteins enabling the synthetic lethality with MYC and AURKB inhibition may be encoded by a subset of these centrosomal genes or even the cumulative effect of upregulating many genes that alter centrosome function, as MYC appears to do. Furthermore, the CPPC targets AURKB activity to the inner centromere where it plays a key role in regulating spindle microtubule attachment and the integrity of the spindle assembly checkpoint (SAC) [17]. Since, knockdown with inhibitory RNAs of any of AURKB, INCENP or Survivin, all components of the CPPC, produces a synthetic lethal effect with MYC [14], one might speculate that the overdependence of MYC expressing cells on robust CPPC function relates to proper kinetochore integrity and spindle assembly in conjunction with the centrosome, which would be enabled by a functional SAC.
However, events arising post chromosome segregation may also play a role in the synthetic lethality of MYC overexpressing tumors to AURKB inhibition. The in vivo treatment of genetically engineered murine models (GEMMs) of both T and B cell lymphoma with the pan- AURK inhibitor, VX-680, leads not just to apoptosis, but also the emergence of polyploidy and induction of autophagy [14]. So, it would seem that in vivo, some VX- 680 treated cells do succeed in completing anaphase, but fail to complete cytokinesis, therefore becoming multinucleate. If and how long these cells can persist in vivo, their contribution to tumor evolution and selective means for targeting their removal are all active areas of investigation.
Isogenic Cell Lines for the Study of MYCVX-680 Synthetic Lethality
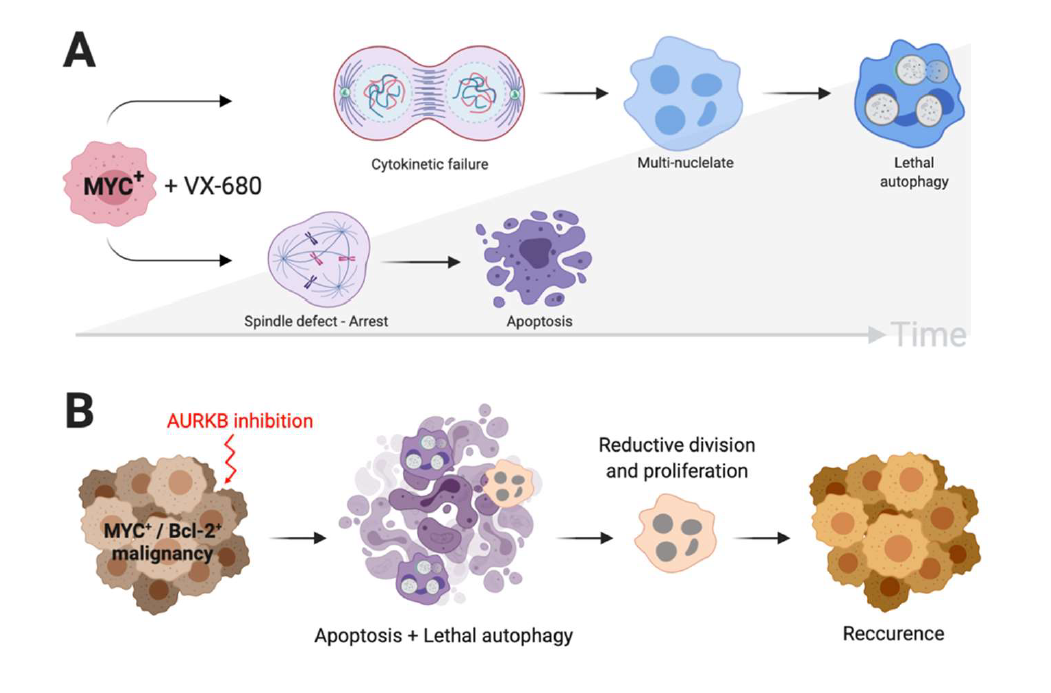
Human retinal pigment epithelial (RPE) cell lines have proven a valuable tool for studying synthetic lethal interactions with MYC [8,14,18]. RPE cells immortalized by the ectopic expression of hTERT are relatively refractory to aurora kinase inhibition. However, adding ectopic expression of MYC (RPE-MYC cells) confers robust sensitivity to these cells. After just 3 days of a transient VX- 680 treatment, RPE-MYC cells are induced to undergo two distinct types of cell death (Figure 1A) [14]. The first to arise occurs in approximately 30-40% of cells and is an apoptotic cell death that follows arrest of cells in pro-metaphase. The BH3-only protein, BCL2L11 (BIM) contributes to events that lead eventually to the cleavage of effector caspases and PARP. However, not all RPE-MYC cells succumb to early apoptosis. Some cells complete mitosis but fail to complete cytokinesis. Polyploidy results and these multinucleate cells come to dominate the culture dish for a short period before they too succumb to cell death. This cell death is not apoptosis, however. The wave of cell death of polyploid cells is associated with a robust induction of autophagy. The autophagy-associated cell death is close to absolute in polyploid RPE-MYC cells and it is this high sensitivity that lends this cell system to the search for modifying events. Escape and the recommencement of reproductive mitosis occurs in approximately 10 in one million RPE-MYC cells [19]. The escape appears to occur through commencement of multipolar cell divisions that reduce the number of nuclei, as the emergent RPE-MYC colonies contain diploid cells with a cell size and nuclear morphology resembling naïve parental cells prior to treatment with VX-680. It is unknown if these cells have acquired further genomic or epigenomic changes and escaping cells are not resistant to re-challenge with VX-680, so the emergence of posttreatment RPE-MYC colonies is not associated with loss of drug response.
A Survey for Modifiers of MYC-VX-680 Synthetic Lethality
Various human cancer-derived cell lines also respond to transient VX-680 treatment with the same two phases of cell death, early apoptosis and autophagy-associated death of polyploid cells [14]. However, there is a differential ability to resume cell proliferation in different human cancer cells, not the near absolute loss of this ability as in RPE-MYC cells. One hypothesis as to why this could be the case is that additional oncogenic events that occur alongside MYC deregulation confer some degree of resistance. This has, until now, not been directly tested. Given the important role that polyploid cells have been ascribed in cancer evolution [20], this is an important line of investigation not only with respect to AURK inhibitor treatment, but for many other therapeutic approaches as well where polyploid cells are induced.
In order to investigate if the oncogenes and tumor suppressor genes that are often altered alongside MYC in cancer can influence the VX-680 phenotype, we constructed a series of isogenic RPE-MYC cell lines. Cells were engineered to overexpress oncogenes and dominant negative forms of tumor suppressors alongside MYC [19]. Then, a transient VX-680 treatment assay was carried out to measure the effects of these manipulations on MYCVX- 680 synthetic lethality. One strength of this approach was that it is an assay of both forms of cell death in response to AURKB inhibition. Apoptosis was assayed after 3 days of VX-680 treatment and polyploid cell survival was assayed 3 days after this.
Pro-survival Bcl2-family Members Prolong Polyploid Cell Survival
Several unexpected findings arose from the study of MYC-VX-680 synthetic lethality in the isogenic cell series.
The tumor suppressor p53 is mutated in many cancers where MYC is overexpressed and MYC-induced apoptosis can be both dependent and independent of p53 activation, depending on context [21]. However, previous work has linked p53 function to Aurora kinases [22,23], so one might expect that a dominant negative version of p53 might disrupt the apoptotic response to VX-680 in RPEMYC cells. This did not happen as the mitotic arrest and apoptosis of diploid RPE-MYC cells was not significantly altered with expression of a dominant negative form of p53, even though this dominant negative p53 did interrupt the apoptotic response to doxorubicin. In RPE-MYC cells, the wave of apoptotic cell death contributing to MYC-VX-680 synthetic lethality does not appear to be p53-dependent.
The mitochondrial apoptotic cascade ultimately leads to the activation of the pro-apoptotic Bcl-2 family proteins Bax, Bak or Bok, mitochondrial outer membrane permeabilization, cytochrome C release and the activation of effector caspases [24]. Another finding of interest revealed by the isogenic cell series was the surprising degree to which VX-680-induced apoptosis in MYC expressing cells was refractory to manipulations that have been found to inhibit MYC-induced apoptosis in other systems, especially the overexpression of pro-survival Bcl-2 family proteins. Overexpression of either Bcl-2 or Bcl-xL was not able to alter the percentage of cells that succumbed to apoptosis after 3 days of VX-680 treatment [19]. One can conclude from this that the cascade of events that leads to apoptotic cell death in VX-680 treated RPEMYC cells is distinct from apoptosis mediated by MYC in other contexts. For example, MYC overexpressing cells will undergo apoptosis upon loss of growth factor stimulation by serum withdrawal, and this can be blocked by Bcl-2 overexpression [25,26]. In addition, MYC-CDK1 synthetic lethal apoptosis can be blocked by Bcl-2 co-expression in Rat1a cells [8]. Therefore, the MYC-VX-680 synthetic lethal apoptosis appears to be distinct from MYC-mediated apoptosis in other contexts.
Perhaps most intriguing was that even though the proapoptotic Bcl-2 family proteins were not able to alter the course of the apoptotic cell death, both Bcl-2 and BclxL were able to prolong the survival of polyploid cells following VX-680 treatment [19]. Polyploid RPE-MYC, engineered to express either of the two pro-survival Bcl-2 proteins, even came to form a monolayer that covered the culture dish following treatment. Eventually, these cells did succumb to autophagy-associated cell death, despite substantial delay of this eventuality. In addition, the emergence of colonies in long-term culture was enabled by Bcl-2 approximately 3-fold. This suggests that inhibition of the autophagy-associated death of polyploid cells may also enable these cells to eventually undergo reductive divisions and resume proliferation, a mechanism that may impact cancer recurrence following treatment (Figure 1B).
Although the mechanism by which Bcl-2 and Bcl-xL enable survival of polyploid RPE-MYC cells following VX-680 treatment does not involve the inhibition of mitochondrial apoptosis, it was found that interaction with a BH3-only protein localized to the endoplasmic reticulum mediates this ability. Beclin 1 (BECN1, ATG6), is a well-described binding partner for Bcl-2 family prosurvival proteins in this organelle [27-29]. The Bcl-2G145A mutant, which lacks the ability to bind Beclin 1, was not able to repress autophagy. Either was a Bcl-2 fusion protein targeted solely to mitochondria. Apparently, the Bcl-2 / Beclin 1 interaction at the endoplasmic reticulum is the required activity.
Implications for AURKB-inhibitory Therapy
The combination of AURK inhibitor plus a BH3 mimetic drug is one that holds promise. In breast cancer cell lines, VX-680 and the BH3 mimetic ABT-737 combine to produce an anticancer effect [30]. Similarly, AURKB inhibition and blockade of Bcl-xL function by the BH3 mimetic, ABT-263, produces a robust anti-cancer effect in cell lines from distinct cancers [31]. The effectiveness of combined therapy, however, has been previously attributed solely to enhanced apoptosis. Our findings suggest that the blockade of pro-survival Bcl-2 members from engaging Beclin 1 is an additional mechanism by which combination treatment could work. Combined therapy could overcome the intrinsic resistance conferred by Bcl-2 to autophagyassociated death in certain malignancies to enhance the potency of AURKB inhibition.
Although the BH3-mimetic, Venetoclax (Venclexta®, ABT-199), is used clinically for the treatment of certain hematopoietic cancers, a clinically approved inhibitor of AURKB is not yet available. Toxicities and the lack of adequate efficacy have limited the use of AURKB inhibitors. However, development of rationale combination therapies and predicative biomarkers would certainly allow drug development efforts targeting AURKB to bear fruit. One malignancy where a combined therapy approach might prove successful is diffuse large B-cell lymphoma (DLBCL) where both MYC and Bcl-2 are commonly activated and aneuploid/polyploid cells are suggested to play a role in relapse [32].
We have tested Bcl-2 and Bcl-xL in our assays [19], but Mcl1 and Bfl-1/A1 also interact directly with Beclin 1 [27,33]. Since similar inhibitory actions toward Beclin 1-induced autophagy might be mediated by Mcl1 and Bfl-1/A1, the target specificity of BH3 memetics used in combination therapy will have to be considered. Tissue and cell-type specific interactions with Beclin 1 may differ.
Future Directions
By no means was the isogenic cell screen for modifiers discussed here a comprehensive screen. We tested a limited number of “best-estimate” cancer genes for their ability to modify MYC-VX-680 synthetic lethality. Hundreds of proteins both prepare the cell for and participate in mitosis. Additional proteins that can act as therapeutic targets to promote or enhance synthetic lethality with MYC may be discovered through RNAi or CRISPR/Cas9-based screens. The RPE-MYC screening system, with its combined robust apoptotic response and the induction of polyploidy upon AURKB inhibition, may be ideal for screening new targets and compounds. What may ultimately prove the ideal target for MYC synthetic lethality could be a specific regulator of AURKB solely during early mitosis or alternatively, only during cytokinesis. Although this has not yet been achieved, such specificity might be accomplished by targeting upstream regulators of the CPPC.
References
2. Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor Perspectives in Medicine. 2014 Jun 1;4(6):a014241.
3. De Barrios O, Meler A, Parra M. MYC’s Fine Line Between B Cell Development and Malignancy. Cells. 2020 Feb;9(2):523.
4. Delgado MD, Albajar M, Gomez-Casares MT, Batlle A, León J. MYC oncogene in myeloid neoplasias. Clinical and Translational Oncology. 2013 Feb 1;15(2):87-94.
5. Slack GW, Gascoyne RD. MYC and aggressive B-cell lymphomas. Advances in Anatomic Pathology. 2011 May 1;18(3):219-28.
6. Wang Y, Engels IH, Knee DA, Nasoff M, Deveraux QL, Quon KC. Synthetic lethal targeting of MYC by activation of the DR5 death receptor pathway. Cancer Cell. 2004 May 1;5(5):501-12.
7. Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. The Journal of Cell Biology. 2007 Jul 2;178(1):93-105.
8. Goga A, Yang D, Tward AD, Morgan DO, Bishop JM. Inhibition of CDK1 as a potential therapy for tumors overexpressing MYC. Nature Medicine. 2007 Jul;13(7):820-7.
9. Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, et al. PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression. Nature Medicine. 2016 Nov;22(11):1321-9.
10. Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYCdriven lymphoma cells. Oncogene. 2012 Mar;31(13):1661-72.
11. Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, et al. Exploiting oncogeneinduced replicative stress for the selective killing of Mycdriven tumors. Nature Structural & Molecular Biology. 2011 Dec;18(12):1331.
12. Liu L, Ulbrich J, Müller J, Wüstefeld T, Aeberhard L, Kress TR, et al. Deregulated MYC expression induces dependence upon AMPK-related kinase 5. Nature. 2012 Mar;483(7391):608-12.
13. Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, et al. The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature. 2015 Sep;525(7569):384-8.
14. Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proceedings of the National Academy of Sciences. 2010 Aug 3;107(31):13836-41.
15. Hook KE, Garza SJ, Lira ME, Ching KA, Lee NV, Cao J, et al. An integrated genomic approach to identify predictive biomarkers of response to the aurora kinase inhibitor PF-03814735. Molecular Cancer Therapeutics. 2012 Mar 1;11(3):710-9.
16. Diaz RJ, Golbourn B, Faria C, Picard D, Shih D, Raynaud D, et al. Mechanism of action and therapeutic efficacy of Aurora kinase B inhibition in MYC overexpressing medulloblastoma. Oncotarget. 2015 Feb;6(5):3359.
17. Carmena M, Wheelock M, Funabiki H, Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nature reviews Molecular cell biology. 2012 Dec;13(12):789-803.
18. Rohrberg J, Van de Mark D, Amouzgar M, Lee JV, Taileb M, Corella A, et al. MYC Dysregulates Mitosis, Revealing Cancer Vulnerabilities. Cell Reports. 2020 Mar 10;30(10):3368-82.
19. Zhang J, Zhang S, Shi Q, Allen TD, You F, Yang D. The anti-apoptotic proteins Bcl-2 and Bcl-xL suppress Beclin1/Atg6-mediated lethal autophagy in polyploid cells. Experimental Cell Research. 2020 May 27:112112.
20. Pienta KJ, Hammarlund EU, Axelrod R, Amend SR, Brown JS. Convergent evolution, evolving evolvability, and the origins of lethal cancer. Molecular Cancer Research. 2020 Jun 1;18(6):801-10.
21. McMahon SB. MYC and the control of apoptosis. Cold Spring Harbor Perspectives in Medicine. 2014 Jul 1;4(7):a014407.
22. Gizatullin F, Yao Y, Kung V, Harding MW, Loda M, Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer Research. 2006 Aug 1;66(15):7668-77.
23. Sasai K, Treekitkarnmongkol W, Kai K, Katayama H, Sen S. Functional significance of aurora kinases– p53 protein family interactions in cancer. Frontiers in Oncology. 2016 Nov 25;6:247.
24. Hardwick JM, Soane L. Multiple functions of BCL- 2 family proteins. Cold Spring Harbor Perspectives in Biology. 2013 Feb 1;5(2):a008722.
25. Bissonnette RP, Echeverri F, Mahboubi A, Green DR. Apoptotic cell death induced by c-myc is inhibited by bcl-2. Nature. 1992 Oct;359(6395):552-4.
26. Fanidi A, Harrington EA, Evan GI. Cooperative interaction between c-myc and bcl-2 proto-oncogenes. Nature. 1992 Oct;359(6395):554-6.
27. Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007 Nov 26;3(6):561-8.
28. Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/ Bcl-XL. Autophagy. 2007 Jul 16;3(4):374-6.
29. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. The EMBO Journal. 2007 May 16;26(10):2527-39.
30. Choi JE, Woo SM, Min KJ, Kang SH, Lee SJ, Kwon TK. Combined treatment with ABT-737 and VX-680 induces apoptosis in Bcl-2-and c-FLIP-overexpressing breast carcinoma cells. Oncology Reports. 2015 Mar 1;33(3):1395-401.
31. Murai S, Matuszkiewicz J, Okuzono Y, Miya H, De Jong R. Aurora B inhibitor TAK-901 synergizes with BCL-xL inhibition by inducing active BAX in cancer cells. Anticancer Research. 2017 Feb 1;37(2):437-44.
32. Islam S, Paek AL, Hammer M, Rangarajan S, Ruijtenbeek R, Cooke L, et al. Drug-induced aneuploidy and polyploidy is a mechanism of disease relapse in MYC/BCL2-addicted diffuse large B-cell lymphoma. Oncotarget. 2018 Nov 13;9(89):35875.
33. Gross A, Katz SG. Non-apoptotic functions of BCL- 2 family proteins. Cell Death & Differentiation. 2017 Aug;24(8):1348-58.