Abstract
Autophagy is the one of the essential pathways for maintaining homeostasis of cells and plays an important regulatory role in cell survival and death. Tp53-induced glycolysis and apoptosis regulator (TIGAR) is a Tp53 target protein and is not only involved in the regulation of metabolism, cell cycle progression and radiation response, but also plays a role in autophagy. TIGAR regulates autophagy either to promote cell survival or contribute to cell death under diverse cellular environments. More research is required to define the differential roles of TIGAR in autophagy and cell survival under various pathophysiological conditions. In this review, we briefly discussed the influences of TIGAR on cell survival through regulating autophagy in different cell types to deepen the understanding of the relationship between TIGAR-mediated metabolic processes in autophagy and cell survival.
Keywords
TIGAR, Autophagy, Cell survival, Apoptosis, Metabolism
Autophagy and Cell Survival/death
Macroautophagy (hereinafter referred to as autophagy) is a process in which cellular lipids, proteins or organelles are engulfed into double membrane vesicles and fused with lysosomes to form autolysosomes, and then, part of cytoplasmic constituents are degraded by acidic hydrolases to release free amino acids and fatty acids [1]. Therefore, autophagy is an important pathway for energy production during starvation and is regarded as a protective response to metabolic stress [2]. Autophagy is interconnected with multiple signaling pathways and has the ability to regulate other cellular and tissue processes, such as cell apoptosis, proliferation, differentiation, and inflammation, those have been connected to many pathological processes, including neurodegeneration, aging, cancer, infection and inflammatory diseases [3]. Autophagy is considered as a double-edged sword in many pathological conditions [4,5]. The regulated autophagy has shown to provide a prosurvival function in response to nutrient starvation, and autophagic removal of damaged or malfunctional mitochondria can also help block the activation of mitochondrial apoptotic pathways [6]. However, in some circumstances, the massive induction of autophagy or blockade of autophagy flux has shown to contribute to or enhance the process of cell death including programmed cell death and inflammation [5,7]. Our previous study indicated that ischemia/reperfusion–induced excessive autophagy exacerbated neuronal damage, which suggested that inhibition of autophagy is favor for cell survival [8].
The process of autophagy in response to cell death and survival is regulated by multiple pathways. There are three main signal transduction pathways that regulate autophagy are involved in regulating metabolic homeostasis for cell survival. These signaling pathways include the energysensing cascade kinases, such as protein kinase A (PKA), adenosine 5’-monophosphate (AMP)-activated protein kinase (AMPK), and mammalian target of rapamycin (mTOR) [5]. Some apoptotic regulators such as Tumor suppressor protein Tp53, caspases, anti-apoptotic protein Flice inhibitory protein (FLIP), death-associated protein kinase (DAPK), also play important regulatory roles in autophagy. In addition, the insufficient energy caused by excessive activation of poly (ADP-ribose) polymerase (PARP1) and activation of DAPK-Protein Kinase D1 (PKD) pathway also leads to necrosis and autophagy, highlighting the interaction between the two pathways. All these studies highlight the intersections between metabolism, autophagy, and cell death.
The pentose phosphate pathway (PPP), a branch of glycolysis, maintains cellular redox homeostasis and reductive synthesis by producing NADPH. NADPH is important for maintaining the reduced form of glutathione (GSH), which controls the levels of superoxide and hydrogen peroxide. Thus, the proper function of PPP and the level of GSH are essential for maintaining basal autophagy. Reactive oxygen species (ROS) produced by mitochondria contributes significantly to respond to nutrient deprivation or metabolic stress, and also functions as signaling molecules that are involved in numerous pathways regulating cell autophagy [9-11]. The balance of intracellular ROS and GSH has a significant effect on autophagy. The dual nature of autophagy involved in cell survival and cell death has been known since its catabolic pathway was discovered in mammalian cells [12]. By elucidating the regulatory role of autophagy in cell survival and death, we can understand the role of autophagy in maintaining cell homeostasis and regulating cell death and survival thresholds.
TIGAR Plays an Inhibitory Function in Autophagy
An introduction of TIGAR
In 2006, Karim Bensaad and his colleagues reported a new downstream gene of p53, TIGAR, an analogue of 6-Phosphofructo-2-kinase/fructose-2, 6-bisphosphatase (PFKFB), which can reduce intracellular glycolysis by lowering fructose-2,6-bisphosphate levels and inhibit oxidative stress and apoptosis through promoting the PPP [13,14]. TIGAR is not always beneficial to regulate intracellular glucose metabolism. For example, TIGAR impairs fru-2, 6-P2 activity to promote hepatic glucose production and increase insulin resistance in the rat model of alcoholic liver disease [15]. Inhibiting Tp53/ TIGAR axis can reduce hypoxia-induced cardiac myocytes death, which is associated with increased glycolysis [16]. PPP generates NADPH to support antioxidant function, so TIGAR is thought to have antioxidant activity as well. The antioxidant activity of TIGAR is also associated with hexokinase 2 (HK2), which enhances HK2 activity and maintains mitochondrial membrane potential and decreases ROS under hypoxic conditions [17].
Recent studies reported that TIGAR was up-regulated in cells under a variety of stress conditions, such as ischemia, ischemia/reperfusion (I/R), and hypoxia [18-21]. Emerging evidence suggests that TIGAR plays a key role in both physiological and pathological processes [18,22]. TIGAR can reprogram the glucose metabolism pathway from glycolysis to promote neural stem cell differentiation to regulate embryonic brain development [22]. The overexpression of TIGAR plays a neuroprotective role in many neurological diseases [18,23,24], and it has the antioxidant and anti-inflammatory effects in I/R-induced neuronal injury via metabolic regulation [25]. In addition to reducing glycolysis, TIGAR is also generally regarded as an anti-apoptotic gene in response to Tp53-induced cell death. For example, in degenerated human and rat nucleus pulposus (NP) cells, TIGAR expression was increased and might mediate the inhibitory effect on hypoxia-induced ROS production and apoptosis [26]. TIGAR activated by Wnt signaling inhibits intracellular ROS and caspase-3 expression to protect against cisplatin-induced spiral ganglion neuron damage [24]. TIGAR is also highly expressed in a variety of tumors and plays an important role in the metabolism of cancers [27]. However, the ability of TIGAR to promote cell survival depends on cell types and environment. In cells that are highly dependent on glycolytic flux for survival, expression of TIGAR is associated with a decrease in survival rate, rather than increase [13,28].
The regulation of autophagy by TIGAR
TIGAR is a multifunctional protein that is not only involved in the regulation of glucose metabolism, cell cycle progression and radiation response, but also in autophagy [17]. A number of studies have shown that autophagy is inhibited by overexpression of TIGAR and induced by knockdown of TIGAR [8,29-31] and TIGAR-regulated autophagy is closely related to clinical disease. For example, TIGAR inhibits proliferation and migration of pulmonary artery smooth muscle cells (PASMCs) by inhibiting autophagy and ROS, thereby improving hypoxia-induced pulmonary arterial hypertension (PAH) [32]. In the lung tissue of patients with idiopathic pulmonary fibrosis (IPF), induction of TIGAR may be one mechanism that autophagy is not activated to promote pulmonary fibrosis [33]. In addition, TIGAR knockout attenuates cardiac fibrosis and upregulates autophagy during heart failure, even if TIGAR -mediated metabolic alterations were more central to this protective mechanism than autophagic effects [34]. TIGAR was elevated and autophagy markers were reduced in 3-nitropropionic acid (3-NP) – induced Huntington’s disease, while dapagliflozin can reverse locomotor and memory impairment by activating autophagy and affecting other factors [35]. These studies indicated that TIGARmediated autophagy regulation may be an effective target to treat some disease and also suggests that there is a negative correlation between TIGAR and autophagy activity. However, there is a study indicated that inhibition of TIGAR reduced LC3-I to LC3-II conversion after severe renal ischemia/reperfusion injury [20], which suggested TIGAR may promote autophagy in some circumstances. These implying that TIGAR plays a positive or negative role in regulating autophagy depending on cell type and stress levels. However, the molecular mechanism by which TIGAR regulates autophagy remains unclear. So far, the literatures have reported that TIGAR mainly regulates autophagy through three signaling pathways.
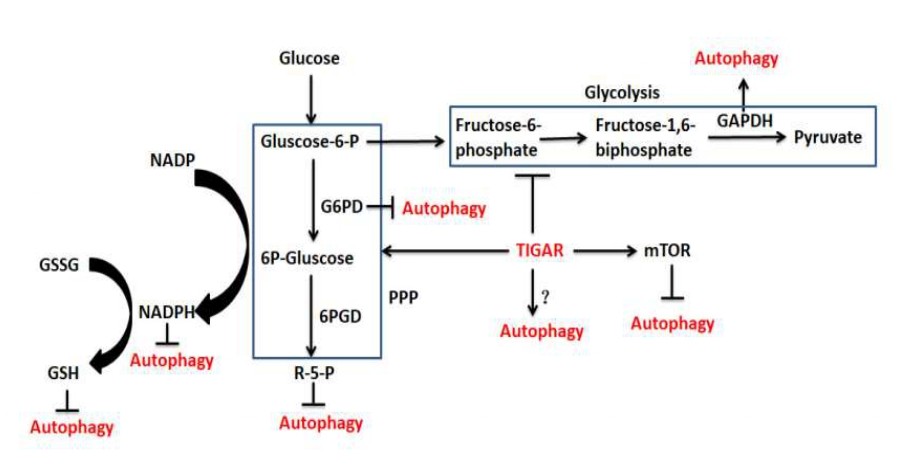
TIGAR regulates autophagy through glycolysis and PPP: In the past several years, it has been found that metabolic processes play important roles in regulating autophagy, which is thought that catabolic processes fortify autophagic flux, whereas anabolic processes may decrease autophagy [36]. Autophagy and cell metabolism are closely related processes. The control of autophagy through metabolic processes can mainly be divided into two ways: one is by directly sensing the levels of metabolic intermediates, and the other is via regulating the oxidative status of cell. Changes in glycolytic enzyme activities can transmit signals to regulate autophagy, such as glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which interacts with mTOR to activate autophagy under low levels of intracellular glucose [37] through translocating to the nucleus and upregulating Atg12 to initiate formation of the autophagosome [38]. mTOR is a major regulator of the autophagic process, and inhibition of mTOR signaling pathway is one of the essential signals to activate autophagy [39]. Recent studies have emphasized that TIGAR knockdown can enhance epriubicin-induced autophagy by inhibiting mTOR pathway [40] and knockdown of TIGAR decreased the phosphorylation levels of mTOR, which was associated with dioscin-induced autophagy [41]. Our recent studies found that overexpression of TIGAR increased the levels of p-mTOR and p-S6KP70, while knockout of TIGAR markedly decreased them in I/R-damaged neurons and cerebral cortex [8], and TIGAR inhibited epirubicin-mediated mTOR pathway activation to exerte a negative impact on autophagy [42]. In addition, Bensaad et al. reported that the modulation of autophagy by TIGAR was not directly mediated by the mTOR signaling pathway, because changes in TIGAR expression did not obviously alter the levels of p70 S6 kinase or the ribosomal S6 protein phosphorylation [43]. This study suggests that TIGAR might modulate autophagy by directly blocking the glycolytic enzyme activity of GAPDH, this needs to be explored further in future studies.
Except for glycolysis, appropriate PPP function and GSH levels are necessary for normal autophagy. PPP is closely related to redox regulation and the production of reductive substances is mediated by PPP in most cells. The PPP catabolizes glucose-6-phosphate to generate reductive substances in the form of NADPH, and in addition, generates ribose-5-phosphate. Enzymes in the PPP are responsive to, and perhaps control, cell survival pathways and autophagy. G6PD, the rate-limiting enzyme in the PPP, generates NADPH, which is the reductant utilized to maintain GSH in its antioxidant reduced form [44]. It has been reported that G6PD inhibition induces autophagy and its overexpression reduces autophagy [45] and TIGAR upregulates G6PD in neuronal cells after brain ischemia/ reperfusion [18]. Thus, whether there is a possibility that TIGAR regulates autophagy by directly affecting G6PD is a research subject in the future. Changes in cellular GSH levels affect autophagic activities in a content dependent manner. Oxidative stress activates genes involved in GSH synthesis and redox status, while higher levels of GSH result in lower oxidative stress in the cell. Therefore, the balance of ROS production and GSH levels may play an important role in regulating the autophagic responses as decreased levels of intracellular GSH leads to autophagy activation [46]. There is a study which implicates that the key regulator of the PPP, ribose-5-phosphate isomerase (RPIA) can regulate autophagy. RPIA inhibits autophagy and LC3 processing through regulation of redox signaling [47]. However, if TIGAR mediates autophagy via regulating RPIA needs to be studied. These observations indicate that the regulation of PPP by TIGAR is important in regulating both the cellular redox status and autophagy (Figure 1).
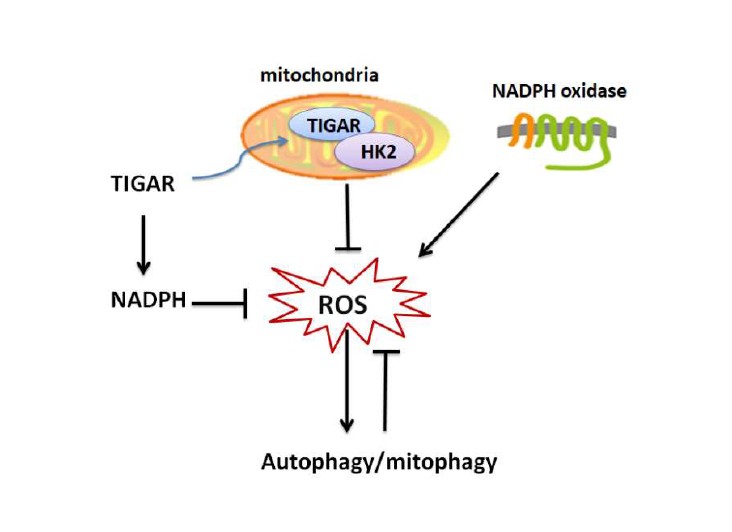
TIGAR regulates autophagy through ROS: ROS is the major cellular signaling molecule to regulate autophagy and in turn, also contributes to reduce ROS [48]. Mitochondria and the NADPH oxidase (NOX) are the two major sources for ROS production in cells. The molecular regulatory mechanisms of autophagy by ROS can be divided into transcriptional and post-transcriptional regulation, and ROS–TIGAR–autophagy is one of the various molecular signal pathways [48]. Bensaad et al. reported that TIGAR regulating autophagy via modulating ROS levels [43]. Kim’s study [20] reported that the increased expression of TIGAR could lead to decreased production of ROS through increasing NADPH, and TIGAR inhibition attenuated autophagy in severe ischemic kidneys, which also indicated that TIGAR regulation of autophagy is closely related to ROS. Recent studies have shown that under hypoxia TIGAR can translocate into mitochondria and interact with HK2, and then limit mitochondrial ROS [21]. It is reported that TIGAR attenuates mitophagy (an autophagic process) through suppressing ROS levels after cardiac ischemia [16]. Our recent studies also found that TIGAR is localized in the mitochondria and reduces mitochondrial ROS in neuronal cells after ischemia/ reperfusion; overexpression of TIGAR in tg-TIGAR mice reduced ROS levels and suppress autophagy activation, while knockout of TIGAR in ko-TIGAR mice exerts the opposite effects [8,18]. Figure 2 briefly summarized TIGAR’s regulation of autophagy through ROS.
The Role of TIGAR-regulated Autophagy in Cell Destiny
TIGAR has different effects on the regulation of autophagy and cell survival under different metabolic conditions. It may maintain the homeostasis of autophagy by rebuilding compromised autophagy and inhibit excessive autophagy through multiple pathways. It has been proven that TIGAR has a vital regulatory role on autophagy to promote or inhibit cell survival [13]. For example, activation of the Tp53-TIGAR axis suppresses autophagy resulting in impaired mitochondrial integrity and subsequent apoptosis, and ultimately exacerbates heart damage after ischemia [16]. While the upregulated TIGAR increases autophagy to guarantee cell survival after severe renal ischemia reperfusion injury [20].
TIGAR plays an anti-apoptotic role by negatively regulating autophagy under nutrient deficiency or metabolic stress [42,43]. It inhibits autophagy in response to nutrient starvation or metabolic stress during heart failure progression [34]. Inhibition of TIGAR expression in cells exposed to PM 2.5 not only blocked the activation of autophagy, but also impaired vascular endothelial growth factor (VEGF) transcription and protein synthesis, thereby alleviating airway inflammatory responses [49]. Wang’s study found that the expression of TIGAR was reduced in ovulated oocytes from high-fat diet (HFD)-fed mice, and specific deficiency of TIGAR in mouse oocytes led to the enhanced ROS levels and strong activation of autophagy. However, the induction of autophagy in response to lack of TIGAR is a protective response to suppress ROS levels, which indicates that TIGAR regulates oocyte maturation closely associated with autophagy [50]. Li et al. found that knockdown of TIGAR significantly reduced nucleus pulposus cell viability and consequently increased compression stress-induced autophagic cell death [51]. All these results suggested that TIGAR could regulate autophagy to control cell survival. The role of TIGAR in regulating autophagy in cancer cell and neuronal cell destiny is described below.
In cancer cells
It has been known that TIGAR is high expressed in many cancers [27]. It is reported that TIGAR knockdown increased cells apoptosis and autophagic cell death in human breast carcinoma [52], as well as in HepG2 cells [31] and MCF-7 cells [53]. TIGAR was upregulated to protect glutamine-treated MCF7 cells against apoptosis through its anti-apoptotic and anti-autophagic effects [54]. Bensaad’s research also showed that TIGAR decreased intracellular ROS and suppressed excessive autophagy in response to nutrient starvation or metabolic stress, and then inhibits apoptosis to exert a negative effect on cancer therapy [43]. On the other hand, there are studies indicating that blocking the activity of TIGAR can up-regulate autophagy and enhance survival of cancer cells [55]. However, numerous investigations indicate that TIGAR plays a dual role in determining cancer cell fate and its dual action is related to the ability to inhibit both apoptosis and autophagy [30,40,42]. Knockdown of TIGAR can induce apoptosis and autophagy in tumor cells [31]. TIGAR knockdown in anticarcinogen-treated cells resulted in induction of protective autophagy in most of the cells surviving apoptosis, which is largely a defense mechanism to save cells from undergoing apoptosis [40,41]. Xie’s study reported that knockdown of TIGAR reduces tumor formation by increasing ROS production and apoptosis, which was enhanced by inhibiting epirubicin-induced autophagy [42], indicating TIGAR plays a dual role by regulating autophagy and apoptosis. Li et al. also reported that TIGAR plays protective role in tumor cells by inhibiting DNA damage and apoptosis, but TIGAR inhibited autophagy activity and this action is unfavorable to cancer cell survival [30]. These studies suggested that knockdown of TIGAR and simultaneous inhibition of autophagy may enhance the anticancer effect of anticarcinogens in some cells. Altogether, as an important regulator of autophagy and apoptosis, TIGAR’s complex effect on various cancer cells and its role in regulating autophagy in tumors still needs to be further defined.
In neurons
The enhanced glycolysis results in less glucose entering the PPP and reduced glutathione levels, making neurons more sensitive to oxidative stress. Our previous studies found that TIGAR is widely distributed in neurons and increases the PPP to produce NADPH, and thus protects I/R brain damage [13,18]. The role of autophagy in neuronal injury is still controversial. Our study and some other researchers had proven that the overactivated autophagy led to self-attacking and cell death [8,56,57]. In ischemic stroke, glucose-oxygen deprivation induces a large number of neuronal death and excessively activated autophagy, so TIGAR inhibits autophagy and reduces neuronal damage. Previous study indicated that TIGAR inhibited autophagy to protect neurons from I/R injury [8]. Zhou and colleagues [29] reported that overexpression of TIGAR can rescue impaired neuronal autophagy induced by hyperglycemia, thereby reducing neuronal apoptosis. Their study suggested that TIGAR might prevent neuronal apoptosis by regulating autophagic process in hyperglycemia conditions. However, others believe that autophagy is a physiological process, and inhibiting autophagy may induce neuronal apoptosis [58-60]. Liu et al. suggested that upregulation of TIGAR expression may block the autophagy-lysosome pathway to reduce KA-induced excitotoxicity [61]. In the study of Wang et al., low-glucose induced autophagy in brain endothelial cells lead to disruption of tight junctions, and overexpression of TIGAR inhibits autophagy to protect endothelial tight junctions by increasing the NADPH yield during hypoglycemic stress [62]. Altogether, these studies indicate that regulated autophagy may be necessary for the self-repair of neurons, while excessive autophagy may lead to neuron damage. TIGAR’s ability in limiting autophagy activity may prevent over activation of autophagy under pathological conditions and thus may be beneficial to neurons.
Conclusions
Autophagy has the function of generating degradation products and nutrients, and removing damaged macromolecules and organelles to control intracellular quality and maintain cell survival during nutrition deficiency and under stress condition. Autophagy plays multiple roles in cell metabolism, development, and cell death via these two basic functions. TIGAR regulates glucose metabolic pathway and autophagy activity to play different roles in different cells under various pathological conditions, but the mechanism by which TIGAR regulates autophagy is still poorly understood. Unraveling the molecular mechanisms by which TIGAR regulates autophagy and its role in cell survival will be a crucial step for us to understand the functions of TIGAR in controlling cell metabolism and autophagy. It could also be expected to yield more insights into complicated interactions between metabolism and autophagy.
Acknowledgments
This work was supported by the Natural Science Foundation of China (No. 81730092, 81872845), The Natural Science Foundation of Jiangsu Province (BK20180207), Jiangsu Provincial Medical Youth Talent (QNRC2016762), Jiangsu Key Laboratory of Neuropsychiatric Diseases (BM2013003) and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).
References
2. Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. The International Journal of Biochemistry & Cell Biology. 2004;36:2445-62.
3. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42.
4. Levine B, Kroemer G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell. 2019;176:11-42.
5. Yan X, Zhou R, Ma Z. Autophagy-Cell Survival and Death. Advancesin Experimental Medicine and Biology. 2019;1206:667-96.
6. Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF- 1-dependent adaptive metabolic response to hypoxia. Journal of Biological Chemistry. 2008;283:10892-903.
7. Crighton D, Wilkinson S, O’Prey J, Syed N, Smith P, Harrison PR, et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121-34.
8. Zhang DM, Zhang T, Wang MM, Wang XX, Qin YY, Wu J, et al. TIGAR alleviates ischemia/reperfusion-induced autophagy and ischemic brain injury. Free Radical Biology and Medicine. 2019;137:13-23.
9. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749-60.
10. Chen Y, Gibson SB. Is mitochondrial generation of reactive oxygen species a trigger for autophagy? Autophagy. 2008;4:246-8.
11. Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. Journal of Cellular Physiology. 2002;192:1-15.
12. Codogno P, Meijer AJ. Autophagy and signaling: their role in cell survival and cell death. Cell Death & Differentiation. 2005;12 Suppl 2:1509-18.
13. Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107-20.
14. Li H, Jogl G. Structural and biochemical studies of TIGAR (TP53-induced glycolysis and apoptosis regulator). Journal of Biological Chemistry. 2009;284:1748-54.
15. Derdak Z, Lang CH, Villegas KA, Tong M, Mark NM, de la Monte SM, et al. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. Journla of Hepatology. 2011;54:164-72.
16. Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, et al. p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia. The Journal of Molecular and Cellular Cardiology. 2012;52:175-84.
17. Lee P, Vousden KH, Cheung EC. TIGAR, TIGAR, burning bright. Cancer & Metabolism. 2014;2:1.
18. Li M, Sun M, Cao L, Gu JH, Ge J, Chen J, et al. A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. The Journal of Neuroscience. 2014;34:7458-71.
19. Sun M, Li M, Huang Q, Han F, Gu JH, Xie J, et al. Ischemia/reperfusion-induced upregulation of TIGAR in brain is mediated by SP1 and modulated by ROS and hormones involved in glucose metabolism. Neurochemistry International. 2015;80:99-109.
20. Kim J, Devalaraja-Narashimha K, Padanilam BJ. TIGAR regulates glycolysis in ischemic kidney proximal tubules. The American Journal of Physiology-Renal Physiology. 2015;308:F298-308.
21. Cheung EC, Ludwig RL, Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:20491-6.
22. Zhou W, Zhao T, Du J, Ji G, Li X, Ji S, et al. TIGAR promotes neural stem cell differentiation through acetyl- CoA-mediated histone acetylation. Cell Death and Disease. 2019;10:198.
23. Zhou JH, Zhang TT, Song DD, Xia YF, Qin ZH, Sheng R. TIGAR contributes to ischemic tolerance induced by cerebral preconditioning through scavenging of reactive oxygen species and inhibition of apoptosis. Scientific Reports. 2016;6:27096.
24. Liu W, Xu X, Fan Z, Sun G, Han Y, Zhang D, et al. Wnt Signaling Activates TP53-Induced Glycolysis and Apoptosis Regulator and Protects Against Cisplatin- Induced Spiral Ganglion Neuron Damage in the Mouse Cochlea. Antioxidants & Redox Signaling. 2019;30:1389- 410.
25. Chen J, Zhang DM, Feng X, Wang J, Qin YY, Zhang T, et al. TIGAR inhibits ischemia/reperfusion-induced inflammatory response of astrocytes. Neuropharmacology. 2018;131:377-88.
26. Jiang LB, Cao L, Ma YQ, Chen Q, Liang Y, Yuan FL, et al. TIGAR mediates the inhibitory role of hypoxia on ROS production and apoptosis in rat nucleus pulposus cells. Osteoarthritis Cartilage. 2018;26:138-48.
27. Geng J, Yuan X, Wei M, Wu J, Qin ZH. The diverse role of TIGAR in cellular homeostasis and cancer. Free Radical Research. 2018;52:1240-9.
28. Kimata M, Matoba S, Iwai-Kanai E, Nakamura H, Hoshino A, Nakaoka M, et al. p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress. The American Journal of Physiology: Heart and Circulatory Physiology. 2010;299:H1908-16.
29. Zhou W, Yao Y, Li J, Wu D, Zhao M, Yan Z, et al. TIGAR Attenuates High Glucose-Induced Neuronal Apoptosis via an Autophagy Pathway. Frontiers in Molecular Neuroscience. 2019;12:193.
30. Li B, Wang Z, Xie JM, Wang G, Qian LQ, Guan XM, et al. TIGAR knockdown enhanced the anticancer effect of aescin via regulating autophagy and apoptosis in colorectal cancer cells. Acta Pharmaceutica Sinica. 2019;40:111-21.
31. Ye L, Zhao X, Lu J, Qian G, Zheng JC, Ge S. Knockdown of TIGAR by RNA interference induces apoptosis and autophagy in HepG2 hepatocellular carcinoma cells. Biochemical and Biophysical Research Communications. 2013;437:300-6.
32. Yamanaka R, Hoshino A, Fukai K, Urata R, Minami Y, Honda S, et al. TIGAR reduces smooth muscle cell autophagy to prevent pulmonary hypertension. The American Journal of Physiology: Heart and Circulatory Physiology. 2020.
33. Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One. 2012;7:e41394.
34. Okawa Y, Hoshino A, Ariyoshi M, Kaimoto S, Tateishi S, Ono K, et al. Ablation of cardiac TIGAR preserves myocardial energetics and cardiac function in the pressure overload heart failure model. The American Journal of Physiology: Heart and Circulatory Physiology. 2019;316:H1366-H77.
35. El-Sahar AE, Rastanawi AA, El-Yamany MF, Saad MA. Dapagliflozin improves behavioral dysfunction of Huntington's disease in rats via inhibiting apoptosisrelated glycolysis. Life Sciences. 2020;257:118076.
36. Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nature Reviews Molecular Cell Biology. 2015;16:461-72..
37. Lee MN, Ha SH, Kim J, Koh A, Lee CS, Kim JH, et al. Glycolytic flux signals to mTOR through glyceraldehyde- 3-phosphate dehydrogenase-mediated regulation of Rheb. Molecular and Cellular Biology. 2009;29:3991-4001
38. Colell A, Ricci JE, Tait S, Milasta S, Maurer U, Bouchier-Hayes L, et al. GAPDH and autophagy preserve survival after apoptotic cytochrome c release in the absence of caspase activation. Cell. 2007;129:983-97.
39. Yan XT, Sun YS, Ren S, Zhao LC, Liu WC, Chen C, et al. Dietary alpha-Mangostin Provides Protective Effects against Acetaminophen-Induced Hepatotoxicity in Mice via Akt/mTOR-Mediated Inhibition of Autophagy and Apoptosis. International Journal of Molecular Sciences. 2018;19.
40. Kumar B, Iqbal MA, Singh RK, Bamezai RN. Resveratrol inhibits TIGAR to promote ROS induced apoptosis and autophagy. Biochimie. 2015;118:26-35.
41. Mao Z, Han X, Chen D, Xu Y, Xu L, Yin L, et al. Potent effects of dioscin against hepatocellular carcinoma through regulating TP53-induced glycolysis and apoptosis regulator (TIGAR)-mediated apoptosis, autophagy, and DNA damage. British Journal of Pharmacology. 2019;176:919-37.
42. Xie JM, Li B, Yu HP, Gao QG, Li W, Wu HR, et al. TIGAR has a dual role in cancer cell survival through regulating apoptosis and autophagy. Cancer Research. 2014;74:5127-38.
43. Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015-26.
44. Wamelink MM, Struys EA, Jakobs C. The biochemistry, metabolism and inherited defects of the pentose phosphate pathway: a review. Journal of Inherited Metabolic Disease. 2008;31:703-17.
45. Mele L, la Noce M, Paino F, Regad T, Wagner S, Liccardo D, et al. Glucose-6-phosphate dehydrogenase blockade potentiates tyrosine kinase inhibitor effect on breast cancer cells through autophagy perturbation. Journal of Experimental & Clinical Cancer Research. 2019;38:160.
46. Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radical Biology and Medicine. 2013;63:207-21.
47. Heintze J, Costa JR, Weber M, Ketteler R. Ribose 5-phosphate isomerase inhibits LC3 processing and basal autophagy. Cell Signaling. 2016;28:1380-8.
48. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and Autophagy: Interactions and Molecular Regulatory Mechanisms. Cellular and Molecular Neurobiology. 2015;35:615-21.
49. Xu H, Xu X, Wang H, Qimuge A, Liu S, Chen Y, et al. LKB1/p53/TIGAR/autophagy-dependent VEGF expression contributes to PM2.5-induced pulmonary inflammatory responses. Scientific Reports. 2019;9:16600.
50. Wang H, Cheng Q, Li X, Hu F, Han L, Zhang H, et al. Loss of TIGAR Induces Oxidative Stress and Meiotic Defects in Oocytes from Obese Mice. Molecular & Cellular Proteomics. 2018;17:1354-64.
51. Li Z, Shao Z, Chen S, Huang D, Peng Y, Chen S, et al. TIGAR impedes compression-induced intervertebral disc degeneration by suppressing nucleus pulposus cell apoptosis and autophagy. Journal of Cellular Physiology. 2020;235:1780-94.
52. Ma T, Zhang Y, Zhang C, Luo JG, Kong LY. Downregulation of TIGAR sensitizes the antitumor effect of physapubenolide through increasing intracellular ROS levels to trigger apoptosis and autophagosome formation in human breast carcinoma cells. Biochemical Pharmacology. 2017;143:90-106.
53. Cui L, Song Z, Liang B, Jia L, Ma S, Liu X. Radiation induces autophagic cell death via the p53/DRAM signaling pathway in breast cancer cells. Oncology Reports. 2016;35:3639-47.
54. Ko YH, Lin Z, Flomenberg N, Pestell RG, Howell A, Sotgia F, et al. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells: implications for preventing chemotherapy resistance. Cancer Biology & Therapy. 2011;12:1085-97.
55. Dewaele M, Maes H, Agostinis P. ROS-mediated mechanisms of autophagy stimulation and their relevance in cancer therapy. Autophagy. 2010;6:838-54.
56. Uchiyama Y, Koike M, Shibata M. Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy. 2008;4:404-8.
57. Qin AP, Liu CF, Qin YY, Hong LZ, Xu M, Yang L, et al. Autophagy was activated in injured astrocytes and mildly decreased cell survival following glucose and oxygen deprivation and focal cerebral ischemia. Autophagy. 2010;6:738-53.
58. Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy. 2013;9:1321-33.
59. Caccamo A, Ferreira E, Branca C, Oddo S. p62 improves AD-like pathology by increasing autophagy. Molecular Psychiatry. 2017;22:865-73.
60. Li Y, Zhang Y, Wang L, Wang P, Xue Y, Li X, et al. Autophagy impairment mediated by S-nitrosation of ATG4B leads to neurotoxicity in response to hyperglycemia. Autophagy. 2017;13:1145-60.
61. Liu ZQ, Liu N, Huang SS, Lin MM, Qin S, Wu JC, et al. NADPH protects against kainic acid-induced excitotoxicity via autophagy-lysosome pathway in rat striatum and primary cortical neurons. Toxicology. 2020;435:152408.
62. Wang CK, Ahmed MM, Jiang Q, Lu NN, Tan C, Gao YP, et al. Melatonin ameliorates hypoglycemic stress-induced brain endothelial tight junction injury by inhibiting protein nitration of TP53-induced glycolysis and apoptosis regulator. Journal of Pineal Research. 2017;63.