Abstract
NOXA is a critical mediator of stress responses to anticancer drugs. This BH3-only protein sets the apoptotic threshold in cancer cells in response to chemotherapies by counteracting the prosurvival BCL-2 family protein MCL-1. A complex and dynamic network relying on both highly controlled gene transcription activity and protein degradation by proteasome, regulates cellular NOXA levels from low in steady state to rapidly enhanced upon stressful condition. Antimitotics and proteasome inhibitors are powerful anticancer treatments that mainly rely on NOXA activity to trigger death in cancer cells. In addition, in case of antimitotics, a wave of cell death based on NOXA and fueled by an automomous secretome spreads through the tumor, revealing new therapeutic opportunities. By neutralizing MCL-1 prosurvival activity, NOXA induces preferential BCL-2 or BCL-xL cancer cell survival dependencies, that can be exploited therapeutically using BH3 mimetics. Since MCL-1 interfers with tumor response to chemotherapies, promoting NOXA expression or using recently available MCL-1 targeting BH3 mimetics are promising opportunities to improve cancer treatments.
Introduction
Apoptosis, a programmed cell death relying on the cascade activation of caspases, regulates many processes ranging from embryonic development to immune homeostasis, and plays a major role in cancer. Escape from apoptosis is indeed one of the fundamental characteristics of tumor cells that frequently exhibit increased expression of the main prosurvival BCL-2 homologues BCL-2, BCL-xL and/ or MCL-1 contributing to tumor progression or resistance to anticancer treatments [1]. Mitochondria Outer Membrane Permeabilization (MOMP) is a key cellular event in apoptosis as subsequent release of cytochrome-c (cyto-c) from the mitochondrial intermembrane space to cytosol through BAX/BAK pores, promotes apoptosome formation and downstream activation of apoptotic effector caspases. MOMP can also lead to the release of other mitochondrial components including mitochondrial DNA that engage additional inflammatory signalling pathways inhibited by apoptotic caspases [2,3]. BCL-2 family proteins tightly control BAX/BAK-dependent MOM permeability through a dynamic network of protein-protein interactions integrating various cellular stresses and finally dictating life or death decisions and cell fates [4]. Chemotherapies often upregulate expression of proapoptotic BCL-2 homologues in cancer cells, shifting by this way the balanced death/survival signals towards apoptosis as an expected cytotoxic effect. Among the proapoptotic BH3- only proteins of the BCL-2 family, NOXA is unique since in preferentially inhibiting the prosurvival BCL-2 homologue MCL-1, it decreases the protective effect MCL-1 exerts on mitochondrial membranes and transfers MOM integrity surveillance and downstream prevention of caspase activation, mostly to BCL-2 and/or BCL-xL. This was observed in particular during mitotic-related stress after antimitotic treatment or during endoplasmic reticulum (ER) stress induced by proteasome inhibitors, where NOXA was shown to accumulate through transcriptional or post-translational mechanisms, as we detail in this review. Importantly, prosurvival members of BCL-2 family are now valuable anticancer targets thanks to BH3 mimetics that have been recently developped. The BCL-2 targeting BH3 mimetic venetoclax already achieved great success in hematological malignancies, including chronic lymphoid leukemia where leukemic cell survival mostly depends on BCL-2. However, deployment of BH3 mimetics in solid tumors is still challenging owing to more complex intrinsic tumor cell survival dependencies in close relation to their intrinsic heterogeneity and plasticity evolving during tumor progression and therapeutic pressure. Increasing our understanding of how BCL-2 family is regulated in solid tumors and how this can be therapeutically exploited, is thus a priority to restore potent cell death signalling in cancer cells in order to improve cancer treatment. In this field, NOXA manipulation offers relevant opportunities since its powerful capacity to neutralize MCL-1 antiapoptotic functions reveals intrinsic or acquired molecular vulnerabilities in tumors regarding their survival dependencies. In this review, we focus on the role and the regulation of BCL-2 family proteins in triggering cell death upon chemotherapies especially in solid tumors with particular interest in the molecular mechanisms that govern NOXA levels during therapeutic stress responses. Various therapeutic strategies, already applicable or prospective, to optimize MCL-1 neutralization by NOXA, are also discussed to envision how to accurately exploit NOXA natural anticancer potential.
BCL-2 family proteins control MOMP and downstream apoptotic caspase activity
Apoptosis is a massive synthetic lethal process in which sustained caspase activation leads to many cleavage events that work together to drive cell death. Remodeling of lipid exposure on cell membrane, cellular shrinkage and cytoskeletal degradation, nuclear condensation and DNA fragmentation, and finally formation of apoptotic bodies are stereotypic hallmarks of apoptosis. Most if not all result from the activation of apoptotic effector caspases, mainly caspase-3 (and caspase-7), that in its active form cleaves more than 1000 substrates of which only some of them are related to apoptosis [5]. Among them, the Inhibitor of Caspase-Activated DNase (ICAD) relieves the nuclease activity of CAD allowing DNA fragmentation or the flippase/scramblase system responsible for membrane lipids remodeling necessary to apoptotic bodies engulfment by macrophages and immune silencing [6]. Interestingly some of them, such as the pyroptotic effector gasdermin E (GSDME), appear to be specifically activated by caspase-3 (but not by caspase-7) while being decisive in cell death modalities engaged by chemotherapy [7]. Caspase-3 activation occurs downstream either from the extrinsic pathway after activation of death receptors and the subsequent formation of the Death Inducing Signaling Complex (DISC) or from the intrinsic pathway that relies on MOMP onset, cyto-c release and apoptosome formation. In the extrinsic pathway, exogenous ligand binding to the death receptors TNFR, FAS or DR3 triggers activation of initiator caspase-8 in the DISC and to direct activation of downstream executioner caspases-3/7 that orchestrate apoptosis. An amplifying apoptotic loop based on BID cleavage by caspase-8 often occurs, linking the extrinsic apoptotic pathway to the intrinsic one. The intrinsic pathway referred as mitochondrial pathway is executed via MOMP-dependent cyto-c release. Together with APAF-1 and procaspase-9, cytosolic cyto-c allows the assembly of the mutimeric apoptosome resulting in caspase-9 self-cleavage and downstream caspase-9- dependent caspases-3/7 activation and rapid proteolytic cleavage of their substrates [8].
MOMP is frequently viewed as an irreversible step of apoptosis commitment and is tightly kept in check by the BCL-2 family proteins [9]. This proteins’ family encompasses both prosurvival and proapoptotic groups and functions as a rheostat governing cellular life or death decisions through dynamic protein-protein interactions controlled at multiple levels. Among the proapoptotic members, the effectors BAX, BAK or BOK possess 3 to 4 (BCL-2 Homology) BH domains and a C-terminal transmembrane domain that can anchor to MOM and have the capacity after activation (for BAX and BAK) to oligomerize and form pores in the MOM causing its permeabilization and the release of apoptogenic factors including cyto-c. Other proapoptotic BCL-2 family members possess only one BH domain (BH3), these BH3- only proteins are considered either as « direct activators » like BIM, tBID and PUMA, that bind to BAX/BAK with high affinities [4,10] or as « sensitizors » like NOXA, BIK or BAD that bind BAX/BAK with very low efficiency if any [11,12]. The prosurvival members of the BCL-2 family include BCL-2, BCL-xL, MCL-1, BCL-W and BFL- 1. They possess 4 BH domains (BH1-BH4) and present a hydrophobic pocket that binds heterotypic BH3 domains. They prevent MOMP onset in keeping in check BAX/ BAK effectors as well as their activators or sensitizors. Sensitizors induce the release of the BH3-only activators or the effectors BAX/BAK from antiapoptotic proteins and thus indirectly contribute to BAX/BAK activation [10]. Importantly, all these proteins interact through their highly conserved BH3 domain that can embed in the BH3 binding pocket present only in multi-BH-domain proteins. The BH3-only members associate with prosurvival or proapoptotic multidomain proteins with various affinities finally depicting a hierarchy of specific interactions among the BCL-2 family. Their binding on BAX or BAK induces allosteric changes (activation) that promote the BAX/BAK oligomerization and pore formation in the MOM.
At steady state, the BCL-2 family mediates mitochondria integrity in preventing BAX/BAK activation and pore formation in MOM (Figure 1). Under stress conditions, BCL-2 family then orchestrates an adaptative cell fate response leading to either survival or cell death depending on many parameters including stress intensity or cellular context. The stress-integrative function of BCL-2 network indeed relies on an elaborated dialogue between BCL-2 proteins and also with effectors of various intracellular signalling pathways such as cell metabolism, autophagy or DNA damage response [4]. This is highly regulated in a dynamic way and at multi-levels, including transcriptional regulation, post-translational modifications, or subcellular localization. Both the relative abundance and affinities of BCL-2 family proteins have critical importance and differences in either can greatly change cell outcome since it dictates the cellular capacity to activate BAX or BAK effectors within the MOM or to interfere for example with calcium signalling in the endoplasmic reticulum [13]. Numerous post-translational modifications contribute to regulate interactions between BCL-2 family proteins [14], for example the mitotic arrest-induced decrease of BCL-xL affinity for BAX in relation to its phosphorylation on serine 62 residue fosters mitotic death [15].
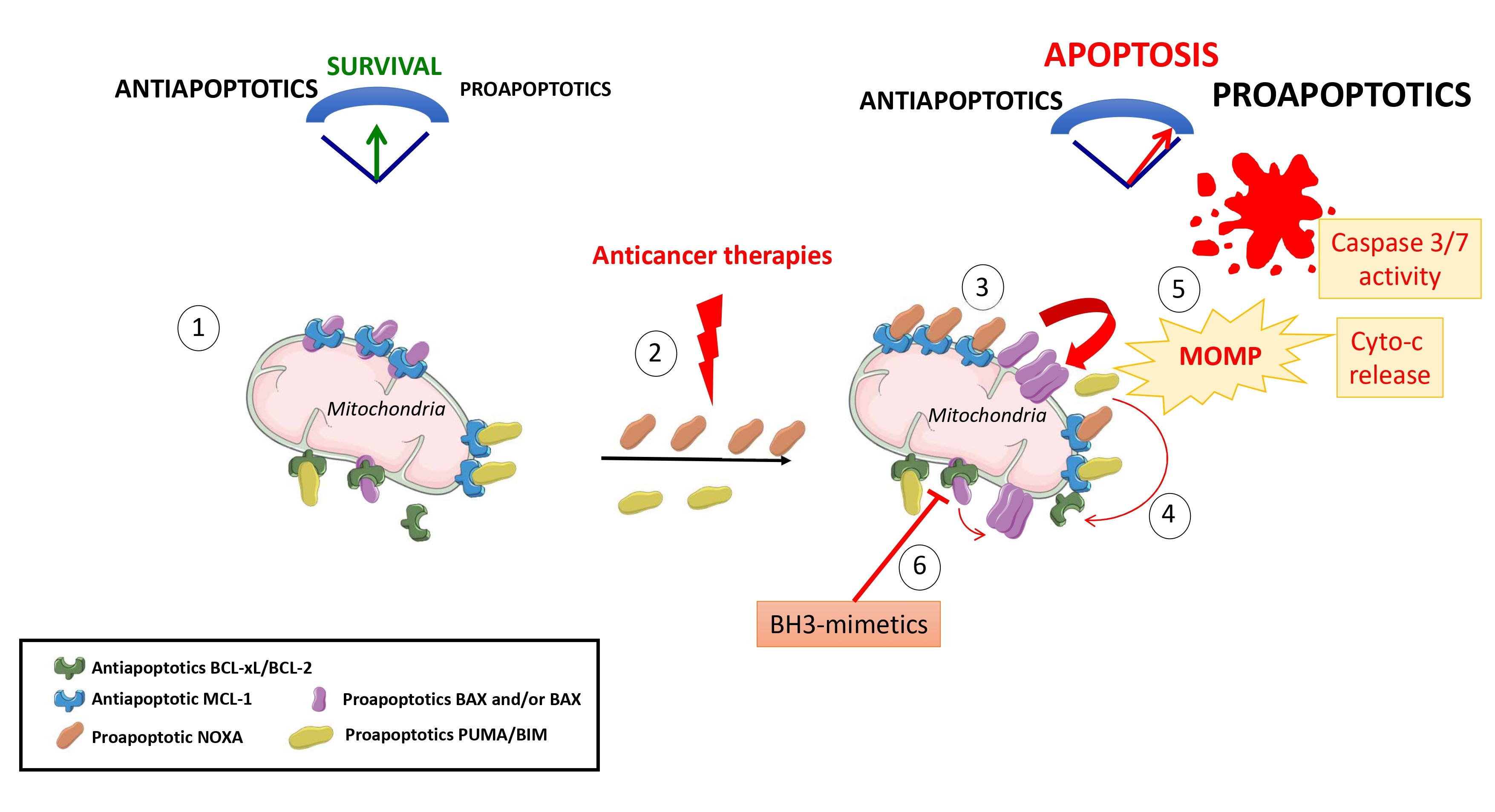
In addition to cyto-c release that is fundamental for apoptotic caspase activition, the inhibitors of IAP (inhibitors of apoptosis proteins) SMAC or HTRA2/OMI and the apoptogenic factors Endo-G and AIF, that all reside in the intermembrane space in basal condition, translocate to the cytosol during MOMP, accelerating apoptosis onset. Importantly, when MOMP associates with permeabilization of mitochondrial inner membrane (MIMP), mitochondrial DNA or RNA spilled in the cytosol engage antiviral immune signalling pathways that are however limited by apoptotic cleavage of caspases substrates involved in this process [2,3,16,17].
Prosurvival BCL-2 proteins are relevant targets in oncology
Intrinsically stressfull tumor environment and therapeutic stress, often fosters expression of BH3-only proteins in cancer cells that overcome this disadvantage in enhancing their endogenous levels of antiapoptotic BCL- 2 proteins: BCL-2, BCL-xL or MCL-1 [18-21]. Cancer cells often display simultaneous high levels of both proapoptotic and survival proteins in still life-sustainable equilibrium (Figure 1) and are thus considered as prone to or primed for apoptosis. In contrast, low expression of apoptotic proteins in many adult vital organs like brain, heart, kidney or liver, protects them from excessive apoptosis sensitivity and offers a clinical window to use BCL-2 family manipulation in cancer treatment [22]. Apoptotic priming can be explored in cancer cells using the BH3 profiling functional assay described by Letai’s laboratory. This in vitro test measures the proximity of cellular mitochondria to the apoptotic threshold and eventually identifies which BH3 are active, after delivering titrated doses of distinct proapoptotic BH3 peptides to mitochondria while monitoring MOMP [23,24]. Of note, patients with highly primed cancers exhibit superior clinical response to chemotherapies, thus suggesting the therapeutic interest to promote apoptotic priming in cancer cells [25]. In addition, using a dynamic version of the BH3 profiling assay to measure mitochondria alteration in cancer cells during in vitro exposure to chemotherapies accurately predicts tumor response to these treatments [26].
Importantly, high apoptotic priming in cancer cells gives rise to intrinsic survival dependencies mainly on BCL-2, BCL-xL or MCL-1 and eventually to codependencies (as BCL-xL and MCL-1 one, as discussed later in this review) due to functional redundancy and compensatory roles among the anti-apoptotic BCL-2 proteins [18,21,27-30]. Great efforts have been thus dedicated to the development of compounds that inhibit prosurvival proteins. Small molecules targeting with high affinity the BH3 binding pocket into the prosurvival proteins BCL-2, BCL-xL or MCL- 1 have been identified and these so called BH3 mimetics in competiting with pro-apoptotic proteins sequestered by prosurvival proteins, potently activate apoptotic cell death by promoting BAX/BAK-dependent MOMP (Figure 1). ABT-199 or venetoclax that specifically targets BCL- 2, was the first BH3 mimetic approved in 2016 by the US Food and Drug Administration for treating chromosomal 17p-deleted BCL2-dependent refractory chronic lymphocytic leukemia (CLL) in monotherapy and has already achieved great success in additional hematological malignancies whose leukemic cells rely on BCL-2 for their survival [31,32]. Preclinical models of difficult-totreat tumors such as triple negative breast cancers or non small cell lung carcinoma, also evidenced synergistic antitumoral effects of the first available dual inhibitors targeting BCL-xL and BCL-2 ABT-737 or navitoclax when used in combination with various chemotherapeutics [19]. Since many tumors (in particular solid tumors) increase BCL-xL expression as a mechanism of intrinsic adaptation to progression or therapeutic pressure, BCL-xL inhibition appeared to be of particular interest in their treatment [33]. We and others indeed reported that antimitotic-treated cancer cells exhibit a strong dependence on BCL-xL for their survival revealing their exquisite sensitivity to BCLxL inhibition [15,34,35]. These observations support the rational to combine antimitotics with BCL-xL inhibitors, eventhough thrombopenia induced by BCL-xL inhibition has to be carefully monitored [36]. Targeting MCL-1 by small molecules was more challenging because of its more complex structure. Finally, the molecules S63845 or AMG176 and derivatives exhibiting high selectivity for MCL-1 over BCL-2 or BCL-xL, have been recently identified and already showed promising results with good tolerance in preclinical studies [37,38].
Combining BH3 mimetics with chemotherapy is under intense clinical investigation in solid tumors but has not achieved significant success so far, compared to hematological malignancies. Survival of cancer cells in carcinoma appears complex and heterogeneous, based on more than one pro-survival BCL-2 family proteins in contrast to hematopoietic malignancies whose survival addiction often relies on a single protein [39]. Overall, we still lack a comprehensive understanding of the regulation of carcinoma survival during tumor progression and upon therapeutic pressure. Predictive biomarkers or functional biological assays (such as BH3 profiling cited above) warranting correct use of BH3 mimetics need to be designed and validated to improve their clinical use in cancer treatment in particular in case of solid tumors.
NOXA, a potent messenger of life or death decision in cancer cells upon chemotherapy
Harnessing intrinsic tumor resources to restore functional apoptotic pathways remains a timely anticancer strategy in which the appropriate recruitment of NOXA can help support improved tumor response to chemotherapy. Since its discovery as a novel phorbol-12-myristate- 13-acetate (PMA) responsive gene in T cells [40], then as a product of p53 genotoxic response illustrating its name related to damage in latin [41], NOXA proved to be crucial in fine-tuning cell death decisions in cancer cells treated by various anticancer drugs [42,43]. In addition to genotoxic anticancer drugs initially reported, the antimitotic agent paclitaxel and the proteasome inhibitor bortezomib both rely on the induction of NOXA expression to trigger apoptotic cell death in cancer cells, through distinct mechanisms involving transcriptional regulation or protein stabilization [44,45]. To the best of our knowledge, NOXA essentially inhibits MCL-1 and is often a rate-limiting BH3-only protein in chemo-induced cell death. Thus, understanding how NOXA is regulated in tumors will help to better harness its therapeutic potential in cancer treatment.
Human PMAIP1 localized on 18q21.23 chromosome, contains 3 exons of which exon 2 is not encoded in NOXA protein, except in two instable (BH3 lacking) variants whose function if any, is unknown [46]. PMAIP1 promoter region that extents over 4 kb, encompasses binding sites for over 40 different transcription factors and co-activators including a bona fide p53 (or p73) response element 195 bp upstream of the transcriptional start site, as initially reported upon genotoxic stress [41,47]. Hypoxia, ER or oncogenic stress also lead to PMAIP1 transcription requiring HIF-1α, E2F1, FOXO3, CREB, MYC, NF-κB, IRF3 or ATF3/4 transcription factors (reviewed in [43,48] (see Figure 2). Interestingly, we reported that following cleavage by apoptotic caspases the E2F1 regulator pRb contributes to amplify the E2F1-dependent NOXA gene transcription and apoptotic response to BH3 mimetics [49]. In the opposite, the polycomb group protein Bmi1 involved in histone and DNA CpG methylation or the highly conserved miR-155 during viral infection have been shown to repress NOXA (at least in mouse T and NK cells respectively) suggesting an epigenetic control of NOXA gene [50,51]. Importantly, NOXA is an integral part of the integrated stress response (ISR), a common stress adaptative pathway, that is primarily a prosurvival program, but driving cell signalling toward cell death when stress is too intense [42]. This cellular stress response consists on both global decrease in protein synthesis via eIF2α phosphorylation and induction of selected genes such as ATF4 whose mRNA is still efficiently translated. This latter facilitates transcriptional upregulation of stress-responsive genes that includes PMAIP1.
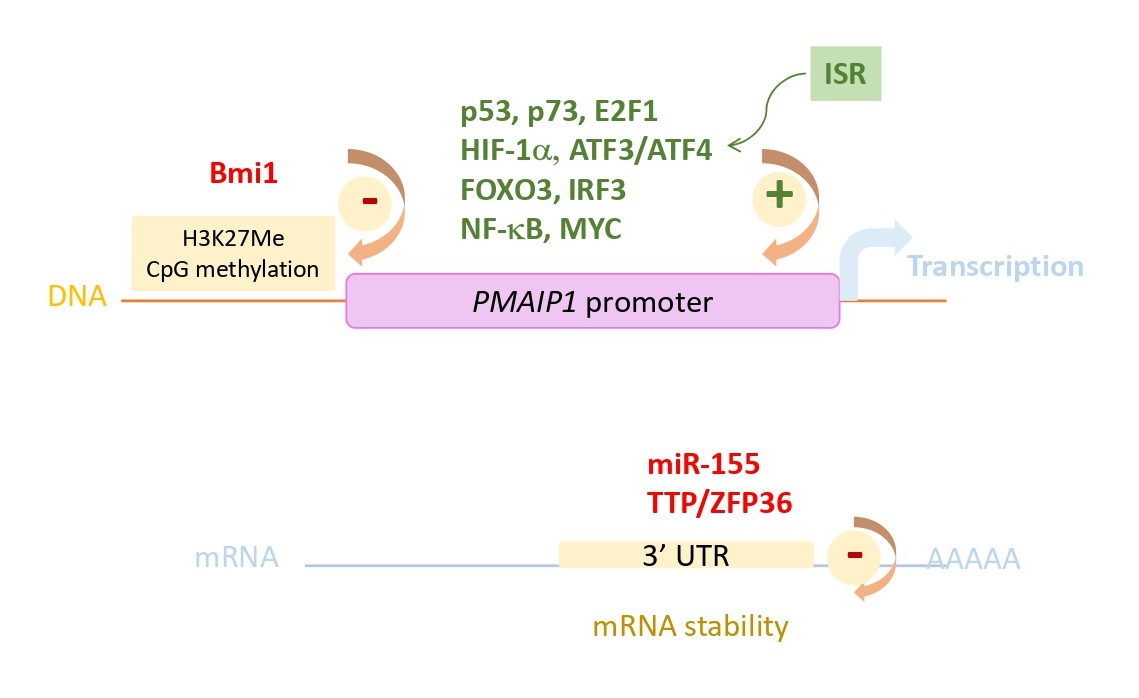
Of note, PMAIP1 gene is rarely mutated in cancers and the sole mutation reported so far displayed no significant difference in cell death induction compared to the wildtype NOXA [52-54]. Some diffuse large B-Cell lymphoma (DLBCL) cell lines harbor genetic co-amplification of PMAIP1 and BCL2 genes that both locate at 18q21, however, this was not observed in primary DLBCL [55].
NOXA is a small protein made up of 54 amino acids (AA) that encompasses a BH3 domain containing the 10–12 shared AA with other BH3-only proteins. Despite lack of secondary structure, this BH3 domain folds to a helical structure when it inserts into the hydrophobic groove of its BCL-2 like binding partner MCL-1, as observed for all BH3 domains [56]. The serine 13 modulates NOXA structure function relationships and its phosphorylation by the kinase CDK5 during glucose stress, that alters NOXA structure, also suppresses its apoptotic function [57,58]. NOXA C-terminal sequence represents a putative mitochondrial targeting domain [59] but more data based on endogenous NOXA protein expression are needed to define its subcellular localization (in particular when NOXA intracellular concentration rises after cellular stress). NOXA preferentially binds MCL-1 through its BH3 domain that embed in the BH3 binding pocket in MCL-1. This binding determines NOXA proapoptotic function since its triple BH3 domain mutant L29E/F32E/ L36E (NOXA-3E) that can no more interact with MCL- 1, is unable to induce apoptosis [60]. As a preferential antagonizer of MCL-1, NOXA competes with, and displaces, other proapoptotic members bound to MCL-1 [61,62] towards BCL-2/BCL-xL or BAX/BAK promoting BAX/BAK pore formation, MOMP and finally apoptosis (Figure 1). Direct interactions between NOXA and BAX or BAK display low affinities compared to that of BIM or tBID (100 fold lesser), indicating that NOXA promotes BAX or BAK activation rather indirectly by freeing BH3-only partners or BAK from MCL-1. For example, NOXA (when highly expressed) cooperates with the activator BIM by displacing it from MCL-1 sequestration, allowing the BIMinducing MOMP and BAX/BAK interactions [63].
Lymphoma cells often express high constitutive levels of NOXA transcript but NOXA protein expression was found to be low, due to rapid degradation relying on a complex proteasome-based degradation process describe below [64]. NOXA protein contains 6 Lysine residues and at least 3 of them can be targeted for ubiquitination and/ or degradation [65,66]. The enzymatic system involved in NOXA ubiquitination is only partially characterized. The multi-unit complexes Skp1-Cullin1-F-box-protein (SCF)-E3 ubiquitin ligases probably contribute to NOXA tagging (including Lysine(K)-48 ubiquitin) for proteasome degradation [67] (Figure 3). The Cullin-RING-ligase-5 (CLR5) E3 ligase when activated by the tricomplex UBE2F/ SAG/CUL5 can in addition modify NOXA by K11 ubiquitin ligation, resulting in its proteasomal targeting. Of note, UBE2F expression in lung cancers correlates with poor patient survival and its knockdown induced spontaneous NOXA accumulation and NOXA-dependent apoptosis, arguing for UBE2F as a potential target and biomarker for patient survival [68]. In the opposite, the K48-dependent ubiquitin hydrolase UCH-L1, protects (specifically among BCL-2 proteins) NOXA from proteosomal degradation and potentiates its genotoxic-induced cell death [69].
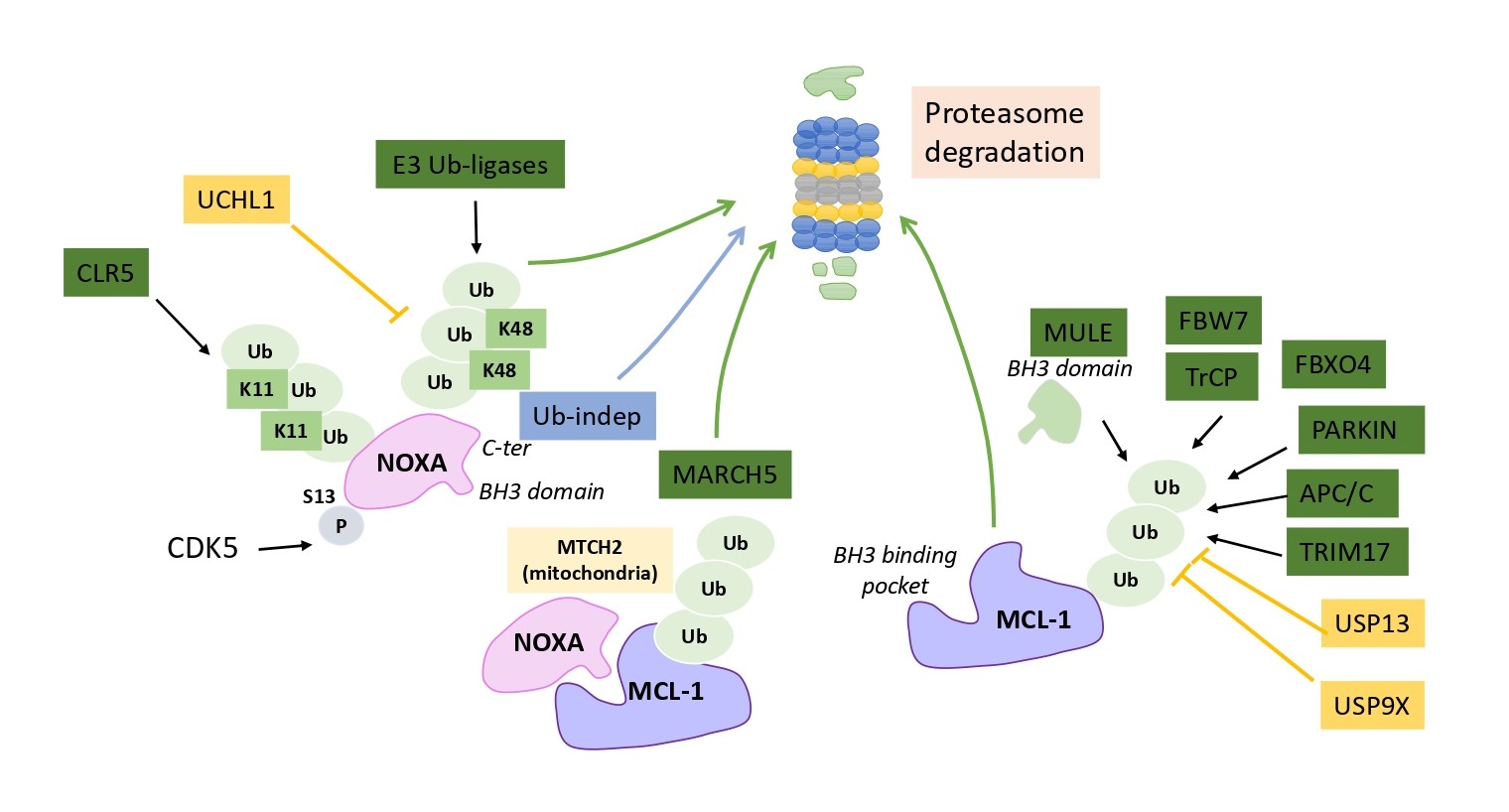
Interestingly, the C-terminal tail (corresponding to its mitochondrial targeting domain) in NOXA serves as a signal for proteasome-dependent but ubiquitinindependent degradation that also controls NOXA stability (as evidenced using K-less NOXA mutants), a process that is probably enhanced when NOXA is not engaged in a complex with MCL-1 [66,70]. Importantly, NOXA binding actively decreases MCL-1 protein half-life in promoting its ubiquitination and subsequent proteasomal degradation [62] (see Figure 3). Intringuingly NOXA is the only BH3- only protein that targets MCL-1 for degradation whereas BIM or PUMA enhances MCL-1 stabilization [71]. The C-terminal portion of NOXA is required for NOXAinduced MCL-1 degradation, as its replacement by residus found in the same position in BIM led to its stabilization [60]. It is not yet clear whether a unique conformational change of MCL-1 induced by NOXA (but not by BIM or PUMA) binding or a specific subcellular localization of the complex MCL-1/NOXA, contributes to MCL-1 ubiquitination but NOXA-induced MCL-1 rapid turnover drives apoptosis in various settings [60,72]. In addition to NOXA engagement, distinct E3 ligases elicited by several cell stress signals are known to ubiquitinate and degrade MCL-1 (Figure 3). They include MULE/HUWE1, SCF-E3 ligases (Skp1-Cullin1-F-box-protein) β-TrCP or FBW7, and also Parkin, MARCH5, APC/C-CDC20 or TRIM17, depending on cellular context, following cell cycle arrest or DNA damage for example [73-75]. Both deubiquitinases USP9X or USP13 remove the polyubiquitin chains that mark MCL-1 for proteasomal degradation enhancing MCL- 1 stability and thus contribute to tumor resistance to BH3 mimetics targeting BCL-2/BCL-xL treatments [76,77]. MCL-1 stability is thus acutely regulated but to what extent its degradation relies on NOXA is still under investigation. Mutations in NOXA BH3 domain that abolish its binding to MCL-1, completely abolished its degrading effect on MCL- 1 [62]. It is possible that this binding competes with the binding of the E3 ligase MULE that also contains a BH3- like domain [78]. In contrast loss of NOXA C-terminal tail that decreases its ubiquitin-independent degradation (and its mitochondrial localization [58]), increases MCL-1 accumulation [66]. The mitochondrial ubiquitin E3 ligase MARCH5 is emerging as a potent inducer of NOXA/MCL- 1 complex degradation and subsequently as a critical factor in cancer cells’ response to BCL-2/BCL-xL targeting BH3 mimetics [79] (Figure 3). Additional proteins such as the MOM adaptator protein MTCH2 contribute to MCL-1/ NOXA complex specific degradation by MARCH5 that needs MCL-1 transmembrane domain and may require a specific orientation of MCL-1/NOXA complex in MOM to operate [80]. Intringuingly, increased MCL-1 in response to MARCH5 loss does not sensitize cancer cells to MCL- 1 inhibitors, but instead sensitizes to BCL-xL inhibition [73]. More studies are needed to better understand how MARCH5 drives cancer cells’ survival dependency and response to BH3 mimetics and how NOXA interplays with this process.
In conclusion, NOXA cellular abundance relies on both tightly-controlled transcriptional regulation that culminates during the integrative cellular stress response and proteasome degradation by still being-described processes. As NOXA strongly monitores MCL-1 survival activity, the NOXA/MCL-1 axis is a major apoptosis rheostat that regulates cell fates in various stressful conditions. This is already well illustrated in cancer cells in particular during mitotic arrest or ER stress as described in the next section.
NOXA shifts cancer cells from MCL-1 and BCL-2/ BCL-xL codependency towards exclusive BCL-xL/ BCL-2 dependency upon anticancer treatments
Defining precisely how cancer cells maintain their survival against the proapoptotic signalling they experienced during oncogenesis or therapies, is of major importance to improve cancer treatment. In many cancer cells, BCL-2/BCL-xL and/or MCL-1 mediate redundant or compensatory functions ensuring apoptosis protection [29,59]. In BCL-2/MCL-1-dependent acute myeloid leukemia (AML) cells, PMAIP1 gene loss confers venetoclax resistance, in contrast the MCL1 one synergizes with the BH3 mimetic to induce cell death [81-83]. In carcinoma cells dependent on BCL-xL and MCL-1 for their survival, expression of NOXA renders these cells dependent on BCL-xL only (Figure 1) [29]. This appears to be crucial in certain circumstances where NOXA-mediated MCL-1 inhibition, is required to trigger cell death as described for example for tumor hyperosmotic stress that reprograms NaCl-challenged cells toward BCL-xL addiction through NOXA induction [84]. In a therapeutic view, ER or mitotic stress inducers are bona fide NOXA-dependent cytotoxic anticancer agents whose combination with BCL-xL targeting BH3 mimetics opens new therapeutic opportunities in solid tumors as emphasized below.
Many reports provide evidence for a critical role of NOXA in the decision between life and death in lymphoma or melanoma cells during treatment by ER stress inducing proteasome inhibitors. Targeting NOXA protein turnover with different inhibitors of the ubiquitinproteasome system like bortezomib, leads to strong accumulation of NOXA protein and induction of NOXAdependent apoptosis in MCL-1-dependent cancer cells [85,86]. The mechanisms for NOXA induction mainly involves enhanced transcription that relies on a p53- independent but cMYC-driven and/or ROS-amplified process [85,87]. The cancer cell response to proteasome inhibitors is reminiscent to the one described as an integrative stress response where ATF4 (a proteasome target) and cMYC (a proteasome substrate) play majors roles [42,87]. It is worthy to note that carcinoma cells that harbor a mesenchymal phenotype often exhibit a higher endogenous level of NOXA compare to epithelial-like ones. This directly relies on ER stress-driven activation of PERK signalling resulting from eIF2α phosphorylation and subsequent induction of ATF3/4-dependent activation of NOXA expression [88,89]. Importantly, this contributes to increased cancer cells’ addiction on BCL-xL leading to their increased sensitivity to BCL-xL inhibition [29,90,91]. Accordingly, epithelial-mesenchymal transition (EMT) induced by E-cadherin coding gene CDH1 knock-down also enhances cell dependency on BCL-xL in relation to PERK-dependent NOXA accumulation that results in NOXA-dependent sensitivity to BCL-xL inhibition [29].
Blocking mitosis progress using antimitotics drugs has proved clinical efficacy in many cancers for decades but optimization of their use still needs to be achieved in refractory tumors. We and others have reported that BCLxL strongly determines cell survival during paclitaxel induced mitotic arrest or Aurora-B inhibitor-mediated polyploidization [15,35,92,93]. We further recently demonstrated that antimitotic agents propagate apoptotic priming across heterogeneously sensitive cancer cells in close relation to NOXA expression induction. NOXA accumulates both in paclitaxel sensitive proliferating cell population through PMAiP1 transcription activation and in insensitive non proliferating ones in relation to the antimitotic-induced secretome produced by mitotic stressed cells. Importantly, both cancer cell populations were committed to NOXA-dependent cell death when submitted to BCL-xL inhibition (in contrast to untreated corresponding cells), revealing the BCL-xL survival addiction they acquired during treatment by either the direct effect of antimitotic drug or the paracrine effect it triggered in tumor (Figure 4). Importantly, this could be therapeutically exploited using a combination of antimitotics and BCL-xL targeting BH3 mimetic that led to better antitumor activity than the antimitotic alone in in vivo experiments where paracrine effects as we evidenced predominate [44]. Using preclinical models, we indeed pointed out that sequential therapeutic schedule consisting on paclitaxel administration then BCL-xL targeting BH3 mimetic, led to a better antitumoral response than the corresponding simultaneous combination. We argue that before dying mitotic stressed cancer cells generate active signals on surrounding cancer cells that in promoting NOXA expression in tumor, drive better tumor response to anticancer treatment.
Mechanistically, antimitotics (mainly paclitaxel) induced in proliferating cancer cells a proapoptotic secretome that relied on the activation of the cytosolic DNA sensor cGAS/ STING immune signalling pathway by paclitaxel-induced micronuclei leading to the secretion of type I interferons (IFN-I) and TNFα (Figure 4) [44]. Increased PMAIP1 gene transcription and NOXA protein accumulation were detected both in mitotic stressed and in cancer cells receiving the paclitaxel-induced secretome (regardless of their TP53 mutation status) however the crucial difference was that STING was mostly required for elaboration of paracrine signals but dispensable for cell autonomous ones. Accordingly, tuning down the Interferon Regulatory Factor 3 (IRF3) or the canonical NF-κB pathway, strongly impaired PMAIP1 transcription, NOXA accumulation and apoptosis priming. In contrast, PMAIP1 gene expression induced during mitotic stressed cells is not yet elucidated. It has been reported that MYC has a potential role in upregulating NOXA (also BIM and BID) and downregulating BCL-xL during mitotic arrest and after mitotic slippage [94]. NOXA seems to accumulate in G2 phase (when canonical protein translation is still active) promoting MCL-1 degradation and BIM release during mitotic arrest [35]. Intringuingly, cGAS can also promote mitotic cell death by suppressing BCL-xL dependent inhibition of MOMP via non transcriptional activity of IRF3 but how this operates is not yet defined [95]. The contribution of the anaphase protease separase in MOMPdependent mitotic cell death, has been recently reported: in cleaving NEK2A-phosphorylated BCL-xL and MCL-1, separase turns them into actors of cell death able to form BAX/BAK-like pores in the MOM [96]. Overall, these findings underscore the erosion of MCL-1 prosurvival function during mitotic stress, that is operated by NOXA and that leads to a switch in survival dependencies in cancer cells from a dual MCL-1/BCL-XL addiction to a BCL-xL preferential one.
NOXA manipulation for therapeutic purpose in oncology
Unleashing the apoptotic potential of NOXA appeared to be essential in cell death induced by antimitotics or proteasome inhibitors as emphasized above, and provides opportunities to improve treatment of refractory tumors. ER stress, ROS production or metabolic changes induced by various types of anticancer drugs, are indirect ways to induce NOXA expression through transcriptional mechanism (Table 1), and as such NOXA could be considered as a decisive part of the apoptotic integrative stress response induced by chemotherapies in cancer cells. This occurs independently of (wild type or mutated) TP53 gene status in cancer cells eventhough in taking part in p53-dependent transcriptional response as originally described, NOXA contributes (like PUMA and BAX) to apoptotic response to chemo-induced genotoxic stress in p53 proficient cancer cells [41]. We previously reported that inhibition of Notch signalling in breast cancer tumors, using γ-secretase inhibitors, led to enhanced PMAIP1 transcription and NOXA-dependent cell death (independently of TP53status) [97]. NOXA gene expression is also actionable by STING activation and this coincids with potent anticancer activity of STING agonists in preclinical models of T lymphoma or breast tumors in monotherapy or combined with BCL-xL inhibition [44,98]. Moreover, pharmacological induction of PMAIP1 can be achieved in low expressing cancer cells using histone deacetylase inhibitors (HDACi) or methyl transferase inhibitors as well as the natural anticancer compound degueulin that by this way forster cancer cell death [55,99].
| Anticancer agents | Mechanism/Function | References |
|---|---|---|
| Cisplatin | DNA Alkylating agent | Tonino 2011 [121], Simonin 2013 [122] |
| Etoposide | Topoisomerase II inhibitor | Shibue 2013 [123] |
| Pemetrexed | DNA synthesis inhibitor | Yan 2014 [124] |
| Paclitaxel | Antimitotic | Lohard 2020 [44] |
| Bortezomib | Proteasome inhibitor/ER stress inducer | Qin 2005 [45], Gomez-Bougie 2007 [86], Armstrong 2010 [125] |
| SNX-275 (Entinostat) | HDAC inhibitor | Zhou 2013 [126] |
| BH3 mimetics | BCL-2/BCL-xL inhibitors | Nechiporuk 2019 [83], Soderqvist 2018 [29] |
| DiABZi (human) CMA (mouse) |
STING agonists | Lohard 2020 [44], Gulen 2017 [98] |
| GSIXII | γ-secretase inhibitor | Seveno 2012 [97] |
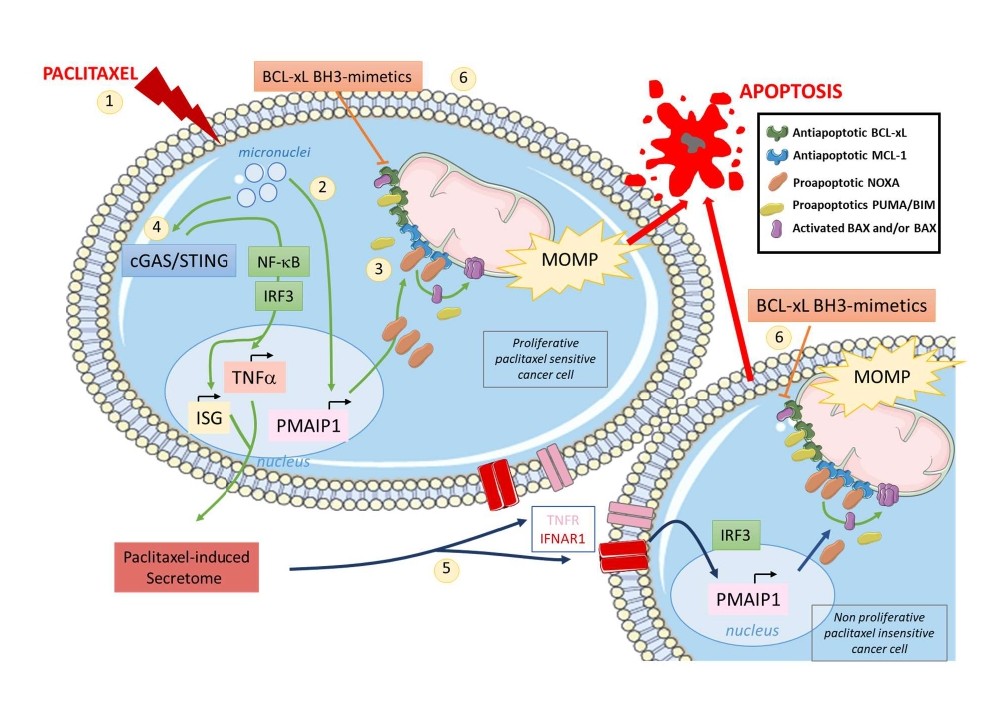
Table 1: Cancer therapies that have been shown to promote cancer cell death by inducing NOXA expression.
Furthermore, interfering with NOXA degradation by proteasome offers new strategies to kill cancer cells. Targeting the ubiquitin ligases CRL (that contribute to NOXA K11 ubiquitination, as described above) by pharmacological inhibition using the neddylation inhibitor MLN4924, or decreasing their expression by gene silencing or using the fatty acid synthase inhibitor orlistat identified during a chemical screen as a potent inducer of NOXA protein stabilization, decreased NOXA turn-over and promoted apoptosis in lymphoma cells and pancreatic tumor growth [67,100,101]. Inhibition of the MOM-associated E3 ligase MARCH5 also led to NOXA accumulation in cancer cells treated by antimitotics or kinase inhibitors and proved to enhance drug-induced cytotoxicity [73,102]. In the same way, derepressing epigenetic control exerted on NOXA deubiquitinase UCH-L1, by HDACi or demethylating agents, increased NOXA stabilization and potentiated chemosensitivity [55,103]. A deeper insight into the ubiquitination/ deubiquitination balance that governs NOXA protein stability may further lead to more novel selective compounds with anticancer potential.
Importantly, tumor adaptative resistance impairing NOXA activity may exist or eventually emerge from anticancer treatments. In this regard, the specific destabilization of NOXA mRNA in melanoma cells has been recently incriminated in resistance to BRAF and EGFR inhibitors involving the MAPK-dependent TTP/ZFP36 activity (Figure 2) [104]. Compensatory mechanisms based on increased expression of prosurvival BCL-2 homologues, can also counteract NOXA proapoptotic activity rendering cancer cells less sensitive to MOMP-dependent apoptosis as observed in lymphoma cells with BCL2 gene amplification or with MCL-1 overexpression [55,82,105] as well as in triple negative breast tumors with MCL1 gene amplification in patients who resist to neoadjuvant chemotherapy [106].
Direct inhibition of MCL-1 has thus emerged as a priority to tackle tumor MOMP resistance. First attempts to target MCL-1 identified compounds that finally failed to directly inhibit MCL-1 in cells, but rather induced the proapoptotic protein NOXA [107]. An NMR-based screen and subsequent structure-guided drug discovery yielded the selective and potent MCL-1 inhibitor S63845 [38]. This compound and derivatives exhibit potent efficacy in vivo in preclinical mouse models of diverse haematological malignancies in monotherapy or in aggressive breast cancer models in combination with taxanes or anti-Her2 targeted therapies, with an acceptable safety margin [108]. The BH3 mimetic AMG176 was identified from a chemical library screen based on MCL-1/BIM disruption and triggered a rapid commitment to apoptosis in models of hematologic malignancies, used alone or in combination with relevant agents, with however dose-dependent reduction in leukocytes after oral administration [37]. Clinical trials testing these MCL-1 inhibitors in humans are still currently underway [109] but an important aspect to take into account is the key pro-survival role of MCL-1 in normal tissues such as heart. MCL-1 is indeed essential for mitochondria homeostasis in murine cardiomyocytes as observed in MCL1 KO mice, raising concerns about potential cardiotoxicity for therapeutics targeting MCL- 1 [110,111]. Whether this activity depends on its BH3 domain and how BH3 mimetics may impact this function is however still undefined. Therefore, careful dosing and schedule are probably needed to mitigate the potential side effects of MCL-1 targeting BH3 mimetics. Another important point is that increased expression of other BCL-2 family prosurvival proteins after MCL-1 targeting, has already been evidenced in cancer cells [73,112]. Of note, the currently available BH3 mimetics that inhibit MCL-1, behave as BIM-like rather than NOXA-like compounds since they enhance MCL-1 accumulation in relation to its decreased degradation. Even if functional inactivation of MCL-1 does not always require its elimination [72], sustained MCL-1 expression in tumors may impact their evolution. It is therefore possible that driving MCL-1 degradation, as mimicking NOXA, might be more efficient than stabilizing its expected inactive form [73]. Interestingly, MCL-1 degradation has been recently achieved using the proteolysis targeting chimera (PROTAC) methodology based on recruitment of a specific E3 ligase to induce proteasome degradation of a target protein after its ubiquitination [113,114]. This approach has the advantage to limit the compound activity in the tissue or tumor that express the chosen E3 ligase but relies on a precise knowledge on which specific E3 ligases are involved in target ubiquitination and where and when they exert this activity.
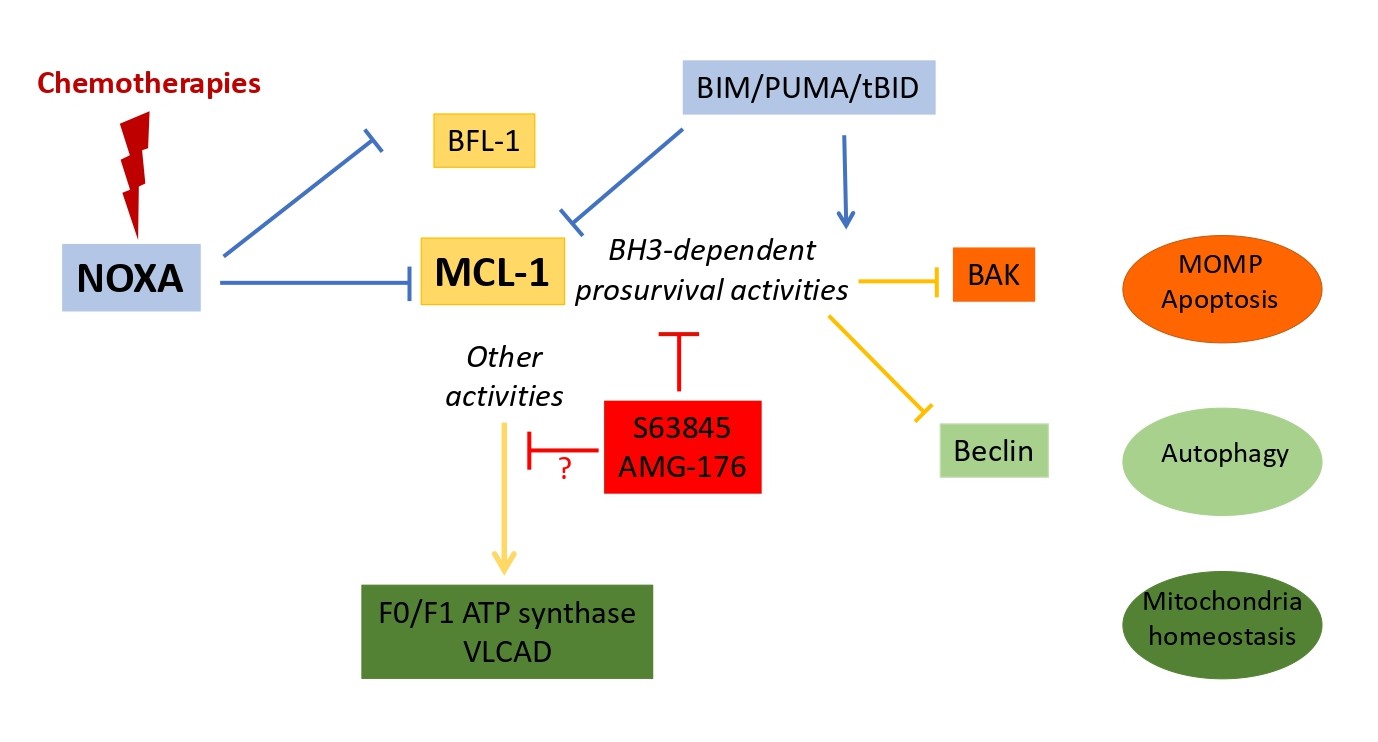
Comparing therapeutic opportunities to target MCL- 1 either through NOXA induction or BH3 mimetics is of particular importance for clinical use since both approaches do not completely overlap. NOXA, in contrast to MCL-1 targeting BH3 mimetics, also binds the prosurvival BCL- 2 homologue BFL-1 (see figure 5) whose contribution in survival of cancer cells eventhough still controversial [115], can drive resistance to BH3 mimetics [29] or to genetic co-dependency with the ataxia-telangiectasia-mutated kinase gene ATM as revealed in some AML cell lines by a genome-scale CRISPR-Cas9 screen [116]. Targeting BFL-1, may be then required for efficient therapeutic intervention in some circumstances and this, in addition to providing the rational to develop potent inhibitors of BFL-1 [117], underpins the interest to promote NOXA activity in anticancer therapies compared to BH3 mimetics. Nevertheless, in cancer cells that display MCL1 amplification, we anticipate that acute inhibition of MCL- 1 using suitable doses of BH3 mimetics, will be probably more easily achieved in patients than indirect activation of NOXA expression. Importantly, mimicking NOXA will also modulate non apoptotic-related BH3-dependent activities of MCL-1 as reported in RAS transformed cells where RASinduced NOXA competes with and liberates Beclin from MCL-1, promoting autophagic cell death and decreased RAS-associated oncogenic activity [118]. Moreover, MCL-1 contributes to mitochondrial homeostasis (as observed in cardiomyocytes) through lipid metabolism or respiration regulation [119,120] but how inhibition of MCL-1 by BH3 mimetics will affect these activities is still unknown (see Figure 5). Further experiments are thus needed to evaluate these crucial points.
Finally, identifying patients who will benefit from NOXA-inducing or MCL-1 targeting therapies is of major importance to advisely use such therapeutic options. Considering the dynamic changes in apoptotic signalling induced by anticancer drugs, it is critical to define which cellular actors support cancer cell survival (ideally at cellular level) during and after acute drug exposure. Functional assays based on cancer cells ex vivo exposure to drugs such as the BH3 profiling assay [26] may aid to predict, at least acute, cytotoxic response to chemotherapies and the best timing for using these drugs.
Conclusion
Restoring functional apoptotic pathways holds considerable therapeutic potential in cancer and in this field, the highly controlled BCL-2 family network is already an actionable target to achieve this objective. As a powerful inhibitor of MCL-1 prosurvival activity, NOXA contributes to commit cell death in response to intense cellular damages including mitotic stress or proteasome inhibition and manipulating its gene transcription and/or protein expression in cancer cells in support of chemotherapy already proved to increase antitumor efficacy. Importantly, NOXA-dependent MCL-1 inhibition often sensitizes solid tumors to BCL-xL inhibition due to their frequent dual MCL-1/BCL-xL survival codependency. However, further mechanistic investigations to elucidate how NOXA levels could be acutely increased in MCL-1-dependent cancer cells, would give more therapeutic opportunities. Inhibition of MCL-1 prosurvival activity using recently available selective BH3 mimetics in combination with chemotherapies or targeted therapies is an attractive strategy for cancer therapy that is currently under clinical evaluation. Of note, these compounds in contrast to NOXAlike molecules, accumulate MCL-1 protein in cancer cells but how this may impact tumor evolution remains to be defined. Importantly, some concerns regarding MCL-1 inhibition in humans are still pending and deserve more dedicated studies: among them, clinical safety since MCL- 1 contributes to normal tissues homeostasis and possibly to MOMP-dependent proinflammatory signalling, or tumor adaptation that will emerge under therapeutic pressure. Identification of patients who will benefit from MCL-1 inhibition using either BH3 mimetics or NOXAinducing therapies (in combination with chemotherapies) is an additional challenge that still needs to be addressed.
Acknowledgments
We thank la Ligue contre le Cancer Grand-Ouest, le Cancéropole Grand-Ouest (CASTHOR network) for their support and all members of the team “stress adaptation and tumor escape” in CRCINA INSERM U1232 for fruitful discussions.
References
2. Rongvaux A, Jackson R, Harman CC, Li T, West AP, De Zoete MR, et al. Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 2014 Dec 18;159(7):1563-77.
3. White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, et al. Apoptotic caspases suppress mtDNAinduced STING-mediated type I IFN production. Cell. 2014 Dec 18;159(7):1549-62.
4. Juin P, Geneste O, Gautier F, Depil S, Campone M. Decoding and unlocking the BCL-2 dependency of cancer cells. Nature Reviews Cancer. 2013 Jul;13(7):455-65.
5. Julien O, Wells JA. Caspases and their substrates. Cell Death & Differentiation. 2017 Aug;24(8):1380-9.
6. Nagata S, Tanaka M. Programmed cell death and the immune system. Nature Reviews Immunology. 2017 May;17(5):333-40.
7. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017 Jul;547(7661):99- 103.
8. Zou H, Li Y, Liu X, Wang X. An APAF-1· cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. Journal of Biological Chemistry. 1999 Apr 23;274(17):11549-56.
9. Goldstein JC, Waterhouse NJ, Juin P, Evan GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant.Nature Cell Biology. 2000 Mar;2(3):156-62.
10. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature Reviews Molecular Cell Biology. 2014 Jan;15(1):49-63.
11. Czabotar PE, Westphal D, Dewson G, Ma S, Hockings C, Fairlie WD, Lee EF, Yao S, Robin AY, Smith BJ, Huang DC. Bax crystal structures reveal how BH3 domains activate Bax and nucleate its oligomerization to induce apoptosis. Cell. 2013 Jan 31;152(3):519-31.
12. Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002 Sep 1;2(3):183-92.
13. Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: changing partners in the dance towards death. Cell Death & Differentiation. 2018 Jan;25(1):65-80.
14. Letai A, Kutuk O. Regulation of Bcl-2 family proteins by posttranslational modifications. Current Molecular Medicine. 2008 Mar 1;8(2):102-18.
15. Bah N, Maillet L, Ryan J, Dubreil S, Gautier F, Letai A, et al. Bcl-xL controls a switch between cell death modes during mitotic arrest. Cell Death & Disease. 2014 Jun;5(6):e1291-.
16. Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature. 2018 Aug;560(7717):238-42.
17. Ning X, Wang Y, Jing M, Sha M, Lv M, Gao P, et al. Apoptotic caspases suppress type I interferon production via the cleavage of cGAS, MAVS, and IRF3. Molecular Cell. 2019 Apr 4;74(1):19-31.
18. Faber AC, Farago AF, Costa C, Dastur A, Gomez- Caraballo M, Robbins R, et al. Assessment of ABT-263 activity across a cancer cell line collection leads to a potent combination therapy for small-cell lung cancer. Proceedings of the National Academy of Sciences. 2015 Mar 17;112(11):E1288-96.
19. Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic?. Cell Death & Differentiation. 2018 Jan;25(1):56-64.
20. Ren D, Tu HC, Kim H, Wang GX, Bean GR, Takeuchi O, et al. BID, BIM, and PUMA are essential for activation of the BAX-and BAK-dependent cell death program. Science. 2010 Dec 3;330(6009):1390-3.
21. Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Molecular Cell. 2013 Sep 26;51(6):751-65.
22. Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, et al. Developmental regulation of mitochondrial apoptosis by c-Myc governs age-and tissuespecific sensitivity to cancer therapeutics. Cancer Cell. 2017 Jan 9;31(1):142-56.
23. Certo M, Moore VD, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006 May 1;9(5):351-65.
24. Ryan J, Montero J, Rocco J, Letai A. iBH3: simple, fixable BH3 profiling to determine apoptotic priming in primary tissue by flow cytometry. Biological Chemistry. 2016 Jul 1;397(7):671-8.
25. Chonghaile TN, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore VD, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011 Nov 25;334(6059):1129-33.
26. Montero J, Sarosiek KA, DeAngelo JD, Maertens O, Ryan J, Ercan D, et al. Drug-induced death signaling strategy rapidly predicts cancer response to chemotherapy. Cell. 2015 Feb 26;160(5):977-89.
27. Goodwin CM, Rossanese OW, Olejniczak ET, Fesik SW. Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer. Cell Death & Differentiation. 2015 Dec;22(12):2098-106.
28. Hata AN, Yeo A, Faber AC, Lifshits E, Chen Z, Cheng KA, et al. Failure to induce apoptosis via BCL-2 family proteins underlies lack of efficacy of combined MEK and PI3K inhibitors for KRAS-mutant lung cancers. Cancer Research. 2014 Jun 1;74(11):3146-56.
29. Soderquist RS, Crawford L, Liu E, Lu M, Agarwal A, Anderson GR, et al. Systematic mapping of BCL-2 gene dependencies in cancer reveals molecular determinants of BH3 mimetic sensitivity. Nature Communications. 2018 Aug 29;9(1):1-3.
30. Xiao Y, Nimmer P, Sheppard GS, Bruncko M, Hessler P, Lu X, et al. MCL-1 is a key determinant of breast cancer cell survival: validation of MCL-1 dependency utilizing a highly selective small molecule inhibitor. Molecular Cancer Therapeutics. 2015 Aug 1;14(8):1837-47.
31. Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with venetoclax in relapsed chronic lymphocytic leukemia. New England Journal of Medicine. 2016 Jan 28;374(4):311-22.
32. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nature Medicine. 2013 Feb;19(2):202-8.
33. Wong M, Tan N, Zha J, Peale FV, Yue P, Fairbrother WJ, et al. Navitoclax (ABT-263) reduces Bcl-xL–mediated chemoresistance in ovarian cancer models. Molecular Cancer Therapeutics. 2012 Apr 1;11(4):1026-35.
34. Gascoigne KE, Taylor SS. Cancer cells display profound intra-and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008 Aug 12;14(2):111- 22.
35. Haschka MD, Soratroi C, Kirschnek S, Häcker G, Hilbe R, Geley S, et al. The NOXA–MCL1–BIM axis defines lifespan on extended mitotic arrest. Nature Communications. 2015 Apr 29;6(1):1-3.
36. Schoenwaelder SM, Jarman KE, Gardiner EE, Hua M, Qiao J, White MJ, et al. Bcl-xL–inhibitory BH3 mimetics can induce a transient thrombocytopathy that undermines the hemostatic function of platelets. Blood, The Journal of the American Society of Hematology. 2011 Aug 11;118(6):1663-74.
37. Caenepeel S, Brown SP, Belmontes B, Moody G, Keegan KS, Chui D, et al. AMG 176, a selective MCL1 inhibitor, is effective in hematologic cancer models alone and in combination with established therapies. Cancer Discovery. 2018 Dec 1;8(12):1582-97.
38. Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016 Oct;538(7626):477-82.
39. Inoue-Yamauchi A, Jeng PS, Kim K, Chen HC, Han S, Ganesan YT, et al. Targeting the differential addiction to anti-apoptotic BCL-2 family for cancer therapy. Nature Communications. 2017 Jul 17;8(1):1-4.
40. Hijikata MA, Kato N, Sato TA, Kagami YO, Shimotohno K. Molecular cloning and characterization of a cDNA for a novel phorbol-12-myristate-13-acetate-responsive gene that is highly expressed in an adult T-cell leukemia cell line. Journal of Virology. 1990 Oct 1;64(10):4632-9.
41. Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita T, et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science. 2000 May 12;288(5468):1053-8.
42. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Reports. 2016 Oct;17(10):1374-95.
43. Ploner C, Kofler R, Villunger A. Noxa: at the tip of the balance between life and death. Oncogene. 2009 Jul 30;27(S1):S84-92.
44. Lohard S, Bourgeois N, Maillet L, Gautier F, Fétiveau A, Lasla H, et al. STING-dependent paracriny shapes apoptotic priming of breast tumors in response to antimitotic treatment. Nature Communications. 2020 Jan 14;11(1):1-6.
45. Qin JZ, Xin H, Sitailo LA, Denning MF, Nickoloff BJ. Enhanced killing of melanoma cells by simultaneously targeting Mcl-1 and NOXA. Cancer Research. 2006 Oct 1;66(19):9636-45.
46. Wang Z, Sun Y. Identification and characterization of two splicing variants of human Noxa. Anticancer research. 2008 May 1;28(3A):1667-74.
47. Grande L, Bretones G, Rosa-Garrido M, Garrido- Martin EM, Hernandez T, Fraile S, et al. Transcription factors Sp1 and p73 control the expression of the proapoptotic protein NOXA in the response of testicular embryonal carcinoma cells to cisplatin. Journal of Biological Chemistry. 2012 Aug 3;287(32):26495-505.
48. Guikema JE, Amiot M, Eldering E. Exploiting the pro-apoptotic function of NOXA as a therapeutic modality in cancer. Expert Opinion on Therapeutic Targets. 2017 Aug 3;21(8):767-79.
49. Bertin-Ciftci J, Barré B, Le Pen J, Maillet L, Couriaud C, Juin P, et al. pRb/E2F-1-mediated caspase-dependent induction of Noxa amplifies the apoptotic effects of the Bcl- 2/Bcl-xL inhibitor ABT-737. Cell Death & Differentiation. 2013 May;20(5):755-64.
50. Yamashita M, Kuwahara M, Suzuki A, Hirahara K, Shinnaksu R, Hosokawa H, et al. Bmi1 regulates memory CD4 T cell survival via repression of the Noxa gene. The Journal of Experimental Medicine. 2008 May 12;205(5):1109-20.
51. Zawislak CL, Beaulieu AM, Loeb GB, Karo J, Canner D, Bezman NA, et al. Stage-specific regulation of natural killer cell homeostasis and response against viral infection by microRNA-155. Proceedings of the National Academy of Sciences. 2013 Apr 23;110(17):6967-72.
52. Jansson AK, Emterling AM, Arbman G, Sun XF. Noxa in colorectal cancer: a study on DNA, mRNA and protein expression. Oncogene. 2003 Jul;22(30):4675-8.
53. Lee SH, Soung YH, Lee JW, Kim HS, Lee JH, Kim HS, et al. Mutational analysis of Noxa gene in human cancers. Apmis. 2003 Jun;111(6):599-604.
54. Mestre-Escorihuela C, Rubio-Moscardo F, Richter JA, Siebert R, Climent J, Fresquet V, et al. Homozygous deletions localize novel tumor suppressor genes in B-cell lymphomas. Blood. 2007 Jan 1;109(1):271-80.
55. Liu Y, Mondello P, Erazo T, Tannan NB, Asgari Z, de Stanchina E, et al. NOXA genetic amplification or pharmacologic induction primes lymphoma cells to BCL2 inhibitor-induced cell death. Proceedings of the National Academy of Sciences. 2018 Nov 20;115(47):12034-9.
56. Kvansakul M, Hinds MG. The structural biology of BH3-only proteins. Methods in Enzymology. 2014 Jan 1; 544:49-74.
57. Karim CB, Espinoza-Fonseca LM, James ZM, Hanse EA, Gaynes JS, Thomas DD, et al. Structural Mechanism for Regulation of Bcl-2 protein Noxa by phosphorylation. Scientific Reports. 2015 Sep 28;5:14557.
58. Lowman XH, McDonnell MA, Kosloske A, Odumade OA, Jenness C, Karim CB, et al. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Molecular Cell. 2010 Dec 10;40(5):823-33.
59. Wilfling F, Weber A, Potthoff S, Vögtle FN, Meisinger C, Paschen SA, et al. BH3-only proteins are tail-anchored in the outer mitochondrial membrane and can initiate the activation of Bax. Cell Death & Differentiation. 2012 Aug;19(8):1328-36
60. Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, et al. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proceedings of the National Academy of Sciences. 2007 Apr 10;104(15):6217- 22.
61. Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nature Reviews Molecular Cell Biology. 2019 Mar;20(3):175-93.
62. Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and BclxL, but not Bcl-2, until displaced by BH3-only proteins. Genes & Development. 2005 Jun 1;19(11):1294-305.
63. Han J, Goldstein LA, Hou W, Rabinowich H. Functional linkage between NOXA and Bim in mitochondrial apoptotic events. Journal of Biological Chemistry. 2007 Jun 1;282(22):16223-31.
64. Dengler MA, Weilbacher A, Gutekunst M, Staiger AM, Vöhringer MC, Horn H, et al. Discrepant NOXA (PMAIP1) transcript and NOXA protein levels: a potential Achilles’ heel in mantle cell lymphoma. Cell Death & Disease. 2014 Jan;5(1):e1013.
65. Baou M, Kohlhaas SL, Butterworth M, Vogler M, Dinsdale D, Walewska R, et al. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica. 2010 Sep 1;95(9):1510-8.
66. Pang X, Zhang J, Lopez H, Wang Y, Li W, O’Neill KL, et al. The carboxyl-terminal tail of Noxa protein regulates the stability of Noxa and Mcl-1. Journal of Biological Chemistry. 2014 Jun 20;289(25):17802-11.
67. Jia L, Yang J, Hao X, Zheng M, He H, Xiong X, et al. Validation of SAG/RBX2/ROC2 E3 ubiquitin ligase as an anticancer and radiosensitizing target. Clinical Cancer Research. 2010 Feb 1;16(3):814-24.
68. Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, et al. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clinical Cancer Research. 2017 Feb 15;23(4):1104-16.
69. Brinkmann K, Zigrino P, Witt A, Schell M, Ackermann L, Broxtermann P, et al. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Reports. 2013 Mar 28;3(3):881-91.
70. Craxton A, Butterworth M, Harper N, Fairall L, Schwabe J, Ciechanover A, et al. NOXA, a sensor of proteasome integrity, is degraded by 26S proteasomes by an ubiquitin-independent pathway that is blocked by MCL-1. Cell Death & Differentiation. 2012 Sep;19(9):1424- 34.
71. Song T, Wang Z, Ji F, Feng Y, Fan Y, Chai G, et al. Deactivation of Mcl-1 by Dual-Function Small-Molecule Inhibitors Targeting the Bcl-2 Homology 3 Domain and Facilitating Mcl-1 Ubiquitination. Angewandte Chemie International Edition. 2016 Nov 7;55(46):14250-6.
72. Lee EF, Czabotar PE, Van Delft MF, Michalak EM, Boyle MJ, Willis SN, et al. A novel BH3 ligand that selectively targets Mcl-1 reveals that apoptosis can proceed without Mcl-1 degradation. Journal of Cell Biology. 2008 Jan 28;180(2):341-55.
73. Arai S, Varkaris A, Nouri M, Chen S, Xie L, Balk SP. MARCH5 mediates NOXA-dependent MCL1 degradation driven by kinase inhibitors and integrated stress response activation. Elife. 2020 Jun 2;9:e54954.
74. Carroll RG, Hollville E, Martin SJ. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Reports. 2014 Nov 20;9(4):1538-53.
75. Harley ME, Allan LA, Sanderson HS, Clarke PR. Phosphorylation of Mcl-1 by CDK1–cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. The EMBO Journal. 2010 Jul 21;29(14):2407-20.
76. Schwickart M, Huang X, Lill JR, Liu J, Ferrando R, French DM, et al. Deubiquitinase USP9X stabilizes MCL1 and promotes tumour cell survival. Nature. 2010 Jan;463(7277):103-7.
77. Zhang S, Zhang M, Jing Y, Yin X, Ma P, Zhang Z, et al. Deubiquitinase USP13 dictates MCL1 stability and sensitivity to BH3 mimetic inhibitors. Nature Communications. 2018 Jan 15;9(1):1-2.
78. Gomez-Bougie P, Ménoret E, Juin P, Dousset C, Pellat- Deceunynck C, Amiot M. Noxa controls Mule-dependent Mcl-1 ubiquitination through the regulation of the Mcl-1/ USP9X interaction. Biochemical and Biophysical Research communications. 2011 Sep 30;413(3):460-4.
79. A. Subramanian, A. Andronache, Y.C. Li, M. Wade, Inhibition of MARCH5 ubiquitin ligase abrogates MCL1- dependent resistance to BH3 mimetics via NOXA. Oncotarget.2016; 7(13):15986-16002.
80. Djajawi TM, Liu L, Gong JN, Huang AS, Luo MJ, Xu Z, et al. MARCH5 requires MTCH2 to coordinate proteasomal turnover of the MCL1: NOXA complex. Cell Death & Differentiation. 2020 Feb 24:1-6.
81. Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discovery. 2019 Jul 1;9(7):890-909.
82. Guièze R, Liu VM, Rosebrock D, Jourdain AA, Hernández-Sánchez M, Zurita AM, et al. Mitochondrial reprogramming underlies resistance to BCL-2 inhibition in lymphoid malignancies. Cancer Cell. 2019 Oct 14;36(4):369-84.
83. Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discovery. 2019 Jul 1;9(7):910-25.
84. Knoll G, Riffelsberger P, Raats D, Kranenburg O, Ehrenschwender M. NOXA-dependent contextual synthetic lethality of BCL-XL inhibition and “osmotic reprogramming” in colorectal cancer. Cell Death & Disease. 2020 Apr 20;11(4):1-1.
85. Pérez-Galán P, Roué G, Villamor N, Montserrat E, Campo E, Colomer D. The proteasome inhibitor bortezomib induces apoptosis in mantle-cell lymphoma through generation of ROS and Noxa activation independent of p53 status. Blood. 2006 Jan 1;107(1):257-64.
86. Gomez-Bougie P, Wuillème-Toumi S, Ménoret E, Trichet V, Robillard N, Philippe M, Bataille R, et al. Noxa up-regulation and Mcl-1 cleavage are associated to apoptosis induction by bortezomib in multiple myeloma. Cancer Research. 2007 Jun 1;67(11):5418-24.
87. Nikiforov MA, Riblett M, Tang WH, Gratchouck V, Zhuang D, Fernandez Y, et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proceedings of the National Academy of Sciences. 2007 Dec 4;104(49):19488-93.
88. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK–eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discovery. 2014 Jun 1;4(6):702-15.
89. Wang Q, Mora-Jensen H, Weniger MA, Perez-Galan P, Wolford C, Hai T, et al. ERAD inhibitors integrate ER stress with an epigenetic mechanism to activate BH3-only protein NOXA in cancer cells. Proceedings of the National Academy of Sciences. 2009 Feb 17;106(7):2200-5.
90. Corcoran RB, Cheng KA, Hata AN, Faber AC, Ebi H, Coffee EM, et al. Synthetic lethal interaction of combined BCL-XL and MEK inhibition promotes tumor regressions in KRAS mutant cancer models. Cancer Cell. 2013 Jan 14;23(1):121-8.
91. Gupta PB, Onder TT, Jiang G, Tao K, Kuperwasser C, Weinberg RA, et al. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 2009 Aug 21;138(4):645-59.
92. Díaz-Martínez LA, Karamysheva ZN, Warrington R, Li B, Wei S, Xie XJ, et al. Genome-wide si RNA screen reveals coupling between mitotic apoptosis and adaptation. The EMBO Journal. 2014 Sep 1;33(17):1960-76.
93. Shah OJ, Lin X, Li L, Huang X, Li J, Anderson MG, et al. Bcl-XL represents a druggable molecular vulnerability during aurora B inhibitor-mediated polyploidization. Proceedings of the National Academy of Sciences. 2010 Jul 13;107(28):12634-9.
94. Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, et al. MYC is a major determinant of mitotic cell fate. Cancer Cell. 2015 Jul 13;28(1):129-40.
95. Zierhut C, Yamaguchi N, Paredes M, Luo JD, Carroll T, Funabiki H. The cytoplasmic DNA sensor cGAS promotes mitotic cell death. Cell. 2019 Jul 11;178(2):302-15.
96. Hellmuth S, Stemmann O. Separase-triggered apoptosis enforces minimal length of mitosis. Nature. 2020 Apr;580(7804):542-7.
97. Séveno C, Loussouarn D, Bréchet S, Campone M, Juin P, Barillé-Nion S. γ-Secretase inhibition promotes cell death, Noxa upregulation, and sensitization to BH3 mimetic ABT-737 in human breast cancer cells. Breast Cancer Research. 2012 Jun 1;14(3):R96.
98. Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, et al. Signalling strength determines proapoptotic functions of STING. Nature Communications. 2017 Sep 5;8(1):1-0.
99. Brodská B, Otevřelová P, Holoubek A. Decitabineinduced apoptosis is derived by Puma and Noxa induction in chronic myeloid leukemia cell line as well as in PBL and is potentiated by SAHA. Molecular and cellular Biochemistry. 2011 Apr 1;350(1-2):71-80.
100. Knowles LM, Axelrod F, Browne CD, Smith JW. A fatty acid synthase blockade induces tumor cell-cycle arrest by down-regulating Skp2. Journal of Biological Chemistry. 2004 Jul 16;279(29):30540-5.
101. Wang Y, Luo Z, Pan Y, Wang W, Zhou X, Jeong LS, et al. Targeting protein neddylation with an NEDD8- activating enzyme inhibitor MLN4924 induced apoptosis or senescence in human lymphoma cells. Cancer Biology & Therapy. 2015 Mar 4;16(3):420-9.
102. Haschka MD, Karbon G, Soratroi C, O’Neill KL, Luo X, Villunger A. MARCH5-dependent degradation of MCL1/ NOXA complexes defines susceptibility to antimitotic drug treatment. Cell Death & Differentiation. 2020 Feb 3:1-6.
103. Brinkmann K, Zigrino P, Witt A, Schell M, Ackermann L, Broxtermann P, et al. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Reports. 2013 Mar 28;3(3):881-91.
104. Montero J, Gstalder C, Kim DJ, Sadowicz D, Miles W, Manos M, et al. Destabilization of NOXA mRNA as a common resistance mechanism to targeted therapies. Nature communications. 2019 Nov 14;10(1):1-5.
105. Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood, The Journal of the American Society of Hematology. 2010 Apr 22;115(16):3304-13.
106. Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discovery. 2014 Feb 1;4(2):232-45.
107. Mallick DJ, Soderquist RS, Bates D, Eastman A. Confounding off-target effects of BH3 mimetics at commonly used concentrations: MIM1, UMI-77, and A-1210477. Cell Death & Disease. 2019 Feb 22;10(3):1-3.
108. Merino D, Whittle JR, Vaillant F, Serrano A, Gong JN, Giner G, et al. Synergistic action of the MCL-1 inhibitor S63845 with current therapies in preclinical models of triple-negative and HER2-amplified breast cancer. Science Translational Medicine. 2017 Aug 2;9(401):eaam7049.
109. Merino D, Kelly GL, Lessene G, Wei AH, Roberts AW, Strasser A. BH3-mimetic drugs: blazing the trail for new cancer medicines. Cancer Cell. 2018 Dec 10;34(6):879-91.
110. Thomas RL, Roberts DJ, Kubli DA, Lee Y, Quinsay MN, Owens JB, et al. Loss of MCL-1 leads to impaired autophagy and rapid development of heart failure. Genes & Development. 2013 Jun 15;27(12):1365-77.
111. Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes & Development. 2013 Jun 15;27(12):1351-64.
112. Seiller C, Maiga S, Touzeau C, Bellanger C, Kervoëlen C, Descamps G, et al. Dual targeting of BCL2 and MCL1 rescues myeloma cells resistant to BCL2 and MCL1 inhibitors associated with the formation of BAX/ BAK hetero-complexes. Cell Death & Disease. 2020 May 5;11(5):1-4.
113. Papatzimas JW, Gorobets E, Maity R, Muniyat MI, MacCallum JL, Neri P, et al. From inhibition to degradation: targeting the antiapoptotic protein myeloid cell leukemia 1 (MCL1). Journal of Medicinal Chemistry. 2019 May 22;62(11):5522-40.
114. Wang Z, He N, Guo Z, Niu C, Song T, Guo Y, et al. Proteolysis targeting chimeras for the selective degradation of Mcl-1/Bcl-2 derived from nonselective target binding ligands. Journal of Medicinal Chemistry. 2019 Aug 7;62(17):8152-63.
115. Lee EF, Harris TJ, Tran S, Evangelista M, Arulananda S, John T, et al. BCL-XL and MCL-1 are the key BCL-2 family proteins in melanoma cell survival. Cell Death & Disease. 2019 Apr 24;10(5):1-4.
116. Guerra RM, Bird GH, Harvey EP, Dharia NV, Korshavn KJ, Prew MS, et al. Precision targeting of BFL- 1/A1 and an ATM co-dependency in human cancer. Cell Reports. 2018 Sep 25;24(13):3393-403.
117. Barile E, Marconi GD, De SK, Baggio C, Gambini L, Salem AF, et al. hBfl-1/hNOXA interaction studies provide new insights on the role of Bfl-1 in cancer cell resistance and for the design of novel anticancer agents. ACS Chemical Biology. 2017 Feb 17;12(2):444-55.
118. Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Molecular Cell. 2011 Apr 8;42(1):23-35.
119. Escudero S, Zaganjor E, Lee S, Mill CP, Morgan AM, Crawford EB, et al. Dynamic regulation of long-chain fatty acid oxidation by a noncanonical interaction between the MCL-1 BH3 helix and VLCAD. Molecular Cell. 2018 Mar 1;69(5):729-43.
120. Perciavalle RM, Stewart DP, Koss B, Lynch J, Milasta S, Bathina M, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nature Cell Biology. 2012 Jun;14(6):575-83.
121. Tonino SH, van Laar J, van Oers MH, Wang JY, Eldering E, Kater AP. ROS-mediated upregulation of Noxa overcomes chemoresistance in chronic lymphocytic leukemia. Oncogene. 2011 Feb;30(6):701-13.
122. Simonin K, N’Diaye M, Lheureux S, Loussouarn C, Dutoit S, Briand M, et al. Platinum compounds sensitize ovarian carcinoma cells to ABT-737 by modulation of the Mcl-1/Noxa axis. Apoptosis. 2013 Apr 1;18(4):492-508.
123. Shibue T, Takeda K, Oda E, Tanaka H, Murasawa H, Takaoka A, et al. Integral role of Noxa in p53-mediated apoptotic response. Genes & Development. 2003 Sep 15;17(18):2233-8.
124. Yan J, Zhong N, Liu G, Chen K, Liu X, Su L, Singhal S. Usp9x-and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death & Disease. 2014 Jul;5(7):e1316.
125. Armstrong JL, Flockhart R, Veal GJ, Lovat PE, Redfern CP. Regulation of endoplasmic reticulum stress-induced cell death by ATF4 in neuroectodermal tumor cells. Journal of Biological Chemistry. 2010 Feb 26;285(9):6091-100.
126. Zhou L, Ruvolo VR, McQueen T, Chen W, Samudio IJ, Conneely O, et al. HDAC inhibition by SNDX-275 (Entinostat) restores expression of silenced leukemiaassociated transcription factors Nur77 and Nor1 and of key pro-apoptotic proteins in AML. Leukemia. 2013 Jun;27(6):1358-68.