Abstract
Alzheimer’s disease is the most common form of dementia with polygenic disposition occurring within various populations. A series of molecular studies indicated that there are number of genes linked to late onset Alzheimer’s disease (AD). In the current study, we examined the contribution of B2-AR (Gly16, Arg), TLR2 (-196TO-174del), PICALM (rs3851179) and BDNF (rs6265) alleles and genotypes, and their relevance in response to Rivastigmine in 150 Iranian AD patients and 150 controls. Genotyping was performed employing Tetra-primer ARMS–PCR and RFLP-PCR methods. Our statistical analysis suggests that A and G alleles of B2-AR show significantly negative (Pc=0.02, RR= 0.65, 95%Cl=0.30-0.94) and positive (Pc=0.02, RR=1.21, 95%Cl=1.06-3.29) associations with familial Alzheimer’s disease (FAD), respectively. Interestingly, after excluding APOE ε4 allele, our results indicated that B2-AR A allele confers more significant protection (Pc=0.006, RR=0.52, 95%Cl=0.33-0.83), while G allele (Pc=0.006, RR=1.90, 95%Cl=1.20- 3.00) and GG genotypes (Pc=0.008, RR= 2.25, 95%Cl=1.26-4.06) provide major susceptibility to AD. We also calculated the clinical relevance of the testing utilizing Prevalence-corrected Positive Predictive Value (PcPPV) formula. The PcPPV of B2-AR A and G alleles were 1.5% and 2.7% respectively in FAD patients. Moreover, the PcPPV for Apo E4 negative AD patients carrying B2-AR GG genotype was 3.5%, meaning that such patients have 3.5% absolute risk for developing AD. Pharmacogenetic analysis of AD patients in a two-year follow-up response to Rivastigmine was also performed. Analysis of Genotype-Related Drug Responses demonstrated that patients carrying B2-AR homozygous GG genotypes were the worst responders to Rivastigmine treatment when compared to total AD patients (baseline) (ΔCDR=0.54). Interestingly, patients with B2-AR homozygous AA genotype, had a best response to the Rivastigmine therapy and decreasing trend in the disease severity and symptoms (ΔCDR= -0.12). These findings suggest that B2-AR A allele acts as a recessive allele in positive response to Rivastigmine treatment. Furthermore, we examined the interactions of the genes related to this study with the genes studied previously in the same patients for response to Rivastigmine. The results indicated, patients, carrying B2-AR AX-A2M AX bigenic genotype show good response (ΔCDR= 0.18) and B2-AR GX- IL6 GX carriers were bad responder to Rivastigmine treatment (ΔCDR= 0.97).
Keywords
Alzheimer, B2-AR, TLR2, PICALM, BDNF, Rivastigmine
Introduction
Alzheimer’s disease (AD) is a devastating neurodegenerative disorder with polygenic and multifactorial inheritance, determined by progressive loss of memory and other cognitive functions. AD is characterized by hallmark pathological changes such as extracellular aggregation of amyloid β (Aβ), intraneuronal neurofibrillary tangles that lead to brain atrophy and loss of neural tissue [1,2]. Alzheimer’s disease is categorized according to the age of onset as early-onset (EOAD) or lateonset AD (LOAD) [3]. And, based on family history, it is classified as sporadic (SAD) or familial Alzheimer’s disease (FAD) [4]. There are various genetic and environmental factors involved in the pathogenesis of AD which makes the etiology of the disease complicated however, testing for genetic markers and genes are useful tools to understand the pathogenesis of wide range of polygenic diseases such as cancers and AD [5-8]. In this regard, several studies were described the association of AD with some polymorphic genes such as Apolipoprotein E (ApoE4), Presenilin 2 (PS2), Alpha-2 macroglobulin (A2M), and Interleukin 6 (IL 6) [5,9,10]. In recent years various genome-wide associations and meta-analysis were performed to find inhibitory and predisposing genes involved in Alzheimer’s. This search resulted in identification of the human genes for B-adrenergic receptor (B2-AR), Toll-like receptor 2 (TLR2), Phosphatidylinositol binding Clathrin assembly protein (PICALM) and brain-derived neurotrophic factor (BDNF) [11-14]. The B2-AR gene is encoded by an intronless gene that is located on chromosome 5q31–5q33. Accumulated convincing evidence from cell culture experiments and animal studies have suggested that β-2-contributes to the AD pathogenesis through its effects on amyloid-β (Aβ) production and complication of inflammation. Several single nucleotide polymorphisms (SNPs) have been described for this gene, notably codon 16 (Gly or Arg). Many studies observed that this SNP might alter cellular trafficking and receptor desensitization [15-17]. Abnormal activation of B2-AR protein might contribute to Aβ accumulation in AD pathogenesis [18,19].
Toll-like receptor 2 (TLR2) is suspected to be the most likely gene associated with the Alzheimer’s disease, since it is localized on chromosome 4q, within the genetic region linked with AD, and that it plays a role in the microglialmediated inflammatory response and amyloid-β (Aβ) clearance [20]. Numbers of studies have shown that TLR2 along with TLR4 and CD14 receptors on the surface of microglial cells are primary receptors for Aβ which upon their interactions stimulate neuronal inflammation and phagocytic initiation. Interestingly, the 196 to -174 polymorphic deletion is shown to change the promoter activity of the TLR2 gene which in turn might have an impact on the development of AD [21,22].
PICALM gene is also known as one of the important candidate genes associated with AD. A strong association of a particular allele of this gene, rs3851179 polymorphism, with AD is demonstrated in number of studies [23,24]. This gene, located on 11qarm of chromosome 14, encodes for 23 different transcripts that are mainly expressed in neurons and shown to play key roles in endocytosis, Iron metabolism and cellular proliferation [25]. PICALM gene contributes to the pathogenesis of AD through intervention in production, transportation, cleaning and other pathways related to Aβ [26].
One of the notable events in the pathogenesis of AD is reduced expression of neurotrophic factors and their receptors, a good example of such factor is Brain-derived neurotrophic factor (BDNF) [27]. The most well-known SNP of BDNF gene is rs6265 that is a conversion of Guanine to Adenine in the 196 nucleotide position (G 196 A), which replaces a Valine for Methionine in prodomain part of the protein structure (Val 66 Met) [28]. This amino acid replacement affects neuronal activity by impairing the secretion of BDNF which is shown to be associated with the establishment of Alzheimer’s disease [29].
Since the first cholinesterase inhibitor production in 1997, cholinergic drugs, such as Rivastigmine, were considered as the initial line of treatment for mild to moderate AD [2,30]. By blocking the acetylcholinesterase enzyme, this drug helps to maintain high levels of acetylcholine in AD patients, a neurotransmitter that plays an important role in human memory and cognitive functions [31]. Innovative studies in the field of pharmacogenetic have shown that differential response of the patients with Alzheimer’s disease to Rivastigmine may be dependent on a particular genotype, hence strengthening the genotype-specific response hypothesis in AD [32]. Therefore, considering the heterogeneity of the disease, and also the importance of the ethnic origin and geographical location of the patients, this study is aimed at investigating the association of B2- AR, TLR2, PICALM, and BDNF alleles and genotypes with AD in Iranian population. In addition, we have investigated the genotype-specific response of Iranian AD patients, with the mentioned polymorphic genotypes to Rivastigmine treatment.
Experimental Procedure
Patients and controls
In the current study, a total of 150 blood samples from unrelated patients with AD (112 SAD and 38 FAD patients, age=7.6 ± 77.73 years) were accumulated from hospitals in Tehran and the Dementia Out-patients Clinic of the Iranian Alzheimer Association (IAA). All these Alzheimer’s disease were genetically independent and diagnosed with following Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) [33]. Criteria and other psychological scales and severity of AD were determined with the Clinical Dementia Rating Sum of Boxes (CDR-SB scale) at the start of treatment by the neurologist. The control group included 150 age, gender, and ethnic background matched healthy individuals, with no family history of dementia or AD that were randomly picked after assessing their cognitive function using the Mini-Mental State Examination [34]. To eliminate possible errors in sample selection and genotyping, the Hardy-Weinberg equilibrium (HWE) test was performed in control group for each polymorphic gene under study. The cognitive functions of participants were assessed by a standard questionnaire form for each patient and controls. Before participating in the research, all individuals gave informed consent. This study was authorized by the ethics committee and review board of Tehran University of Medical Sciences. The severity of AD was measured with CDR-SB after two years, and statistical analysis for the drug response was calculated by the SPSS software version 25 for WINDOWS (Chicago, IL, USA).
B2-AR, TLR2, PICALM, and BDNF genotyping
Blood specimens were gathered in sterile tubes containing EDTA and genomic DNA was extracted using a modified salting-out method. B2-AR polymorphisms (alleles A and G) were detected by polymerase chain reactionrestriction fragment length polymorphism (PCR-RFLP) as described previously [35] according to the following protocol: 5 min denaturation at 94°C then 33 cycles of polymerization each 30 s at 94°C, 30 s at 57°C and 60 s at 72°C, followed by final extension for 5 min at 72°C. The (-196TO-174 del) polymorphism in 5’UTR region of TLR2 gene was determined by Allele-Specific-PCR method as described previously [21]. Briefly, the PCR was performed according to the following protocol: 5 min denaturation at 94°C then 33 cycles of polymerization each for 30 s at 94°C, 30 s at 57°C and 60 s at 72°C followed by 5 min final extension at 72°C. PICALM (rs3851179) genotyping was performed by Allele-Specific-PCR method according to the process reported previously [36]. In order to reduce the cost of BDNF and PICALM genotyping, we designed and employed three and tetra-primer amplification refractory mutation system–polymerase chain (Tetra-primer ARMS–PCR) which are offer fast detection and extreme simplicity at a negligible cost for genotyping, respectively (Table 3). Tetra-primer ARMS–PCR profile of PICALM gene for amplification was 5 min denaturation at 94°C followed by polymerization for 30 s at 94°C, 30 s at 59°C, and 60 s at 72°C for 35 cycles, and 10 min at 72°C for final extension. Genotyping of (rs6265) SNP of BDNF gene by ARMS–PCR was performed with the modified methods reported already [37] which follows 5 min denaturation at 94°C then 33 cycles of polymerization for 30 s at 94°C, 30 s at 57°C, 30 s at 72°C followed by5 min final extension at 72°C. The primer designs were carried out employing Oligo7 program and Ensemble Genome Browser for blasting.
| Gene | RFLP primers |
|---|---|
| B2-AR | B2-AR G:5′-CTT CTT GCT GGC ACC CTA TG-3′ |
| B2-AR A:5′-CTT CTT GCT GGC ACC CTA TA-3′ | |
| PCR primer | |
| TLR2 | TLR2F:5'-CTCGGAGGCAGCGAGAAA-3′ |
| TLR2 R:5'-CTGGGCCGTGCAAAGAAG-3' | |
| Tetra-primer ARMS–PCR | |
| PICALM | PICALMNF:5'-GCAAACAATACACACTTCAGTAAAC-3' |
| PICALM NR:5'-GCTAATTTTTGGTCTTGGTAGAG-3' | |
| PICALM MF:5'-CTATTCAGCTTGGGCGAATTACTAAA-3' | |
| PICALMMR:5'-CACCATATAATAGTTGTGATAGATACC-3' | |
| ARMS–PCR | |
| ΒDNF | BDNF M: CTTTCTCCCTACAGTTCCACCA |
| BDNF N: CATCCAACAGCTCTTCTATCAC | |
| BDNF C: CATCCAACAGCTCTTCTATCAT |
SSP: Specific Sequence Primers; RFLP: Restriction Fragment Length Polymorphism; PCR: Polymerase Chain Reaction; ARMS: Amplification Refractory Mutation System
Table 3: Primers for genotyping of B2-AR, TLR2, PICALM and BDNF genes.
The PCR products were separated by electrophoresis on 6% polyacrylamide gels alongside a 50 bp DNA ladder. The PCR bands were visualized by Ethidium Bromide staining, and the proper size bands for particular alleles were estimated with the corresponding band fragments of the running DNA markers.
Statistical analysis
All data, including clinical and genotyping of the TLR2, PICALM, B2-AR, and BDNF genes, were recorded into a database, and statistical analysis was done using the SPSS version 25 for Windows (Chicago, IL, USA). Genotype and allele frequencies of the TLR2, PICALM, B2-AR, and BDNF were compared between the patients and the controls, and fisher exact tests was used to for statistical analysis of data, and p values were corrected for multiple testing by Bonferoni’s correction. Likewise, Relative risk (odds ratio) was calculated using the Woolf formula: (number of patients with the specific allele/number of patients without this allele)/(number of controls with the specific allele/number of controls without this allele). Prevalence corrected Positive predictive value (PcPPV) evaluates the value of a diagnostic test and indicates the likelihood for a person with a positive test of having or developing the disease, indeed this formula calculate the absolute risk for developing disease. In this case-control study, the following formula was used to determine Prevalence corrected Positive Predictive Value (PcPPV): PcPPV = a/ (a + b) = (PDT+ × PD)/ (PDT+ × PD) + [PCT+ × (1 − PD)].
Where PD is the prevalence of the disease in the general population, PDT+ is the ratio of the patients with a positive test, and PCT+ is the ratio of the controls with a positive test [38].
Results
Distribution of B2-AR, TLR2, PICALM, and BDNF alleles and genotypes in AD Patients
The distribution of B2-AR, TLR2, PICALM and BDNF alleles and genotypes frequencies in the total AD patients and healthy controls are presented in Table 1. The initial data analysis did not show any significant association between the B2-AR alleles or genotypes with total AD patients as compared with the control group (Table 1). But further analysis revealed that the B2-AR G allele is more frequent in the FAD patients (75%) compared to the controls (61.6%) (Pc=0.02), yielding an increased absolute risk of 2.7% (PcPPV=2.7, RR=1.21, 95%CL=1.06-3.29) and On the other hand, the frequency of B2-AR A allele was significantly higher in the control group (38.3%) than in the FAD patients (25%) (Pc=0.02, RR=0.65, 95%CL = 0.30-0.94). Therefore, these results suggest that the B2-AR G and A alleles provided susceptibility for and protection against FAD, respectively.
| All AD Patients | Control | SAD Patients |
FAD Patients |
All AD vs. controls | SAD vs. controls | FAD vs controls | PcPPV% | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Pc | RR | Pc | RR | Pc | RR* | ||||||
| Alleles | |||||||||||
| B2AR | n=300 | n=300 | n=224 | n=76 | |||||||
| A | 105 | 115 | 87 | 19 | NS | NS | 0.02 | 0.65 | 1.5 | ||
| G | 195 | 185 | 137 | 57 | NS | NS | 0.02 | 1.21 | 2.7 | ||
| TLR-2 | n=268 | n=260 | n=194 | n=74 | |||||||
| D | 64 | 61 | 45 | 19 | NS | NS | NS | ||||
| I | 204 | 199 | 149 | 55 | NS | NS | NS | ||||
| PICALM | n=268 | n=260 | n=194 | n=74 | |||||||
| A | 153 | 136 | 110 | 43 | NS | NS | NS | ||||
| G | 115 | 124 | 84 | 31 | NS | NS | NS | ||||
| BDNF | n=224 | n=196 | n=162 | n=62 | |||||||
| A | 36 | 32 | 25 | 11 | NS | NS | NS | ||||
| G | 188 | 164 | 137 | 51 | NS | NS | NS | ||||
| Genotype | |||||||||||
| B2AR | N=150 | N=150 | N=112 | N=38 | |||||||
| A/A | 17 | 18 | 15 | 2 | NS | NS | NS | ||||
| A/G | 71 | 79 | 57 | 15 | NS | NS | NS | ||||
| G/G | 62 | 53 | 40 | 21 | NS | NS | NS | ||||
| TLR-2 | N=134 | N=130 | N=97 | N=37 | |||||||
| D/D | 5 | 5 | 4 | 2 | NS | NS | NS | ||||
| D/I | 52 | 50 | 37 | 15 | NS | NS | NS | ||||
| I/I | 77 | 75 | 56 | 20 | NS | NS | NS | ||||
| PICALM | N=134 | N=130 | N=97 | N=37 | |||||||
| A/A | 40 | 33 | 30 | 11 | NS | NS | NS | ||||
| A/G | 72 | 67 | 50 | 21 | NS | NS | NS | ||||
| G/G | 22 | 30 | 17 | 5 | NS | NS | NS | ||||
| BDNF | N=112 | N=98 | N=81 | N=31 | |||||||
| A/A | 3 | 4 | 2 | 1 | NS | NS | NS | ||||
| A/G | 30 | 24 | 21 | 9 | NS | NS | NS | ||||
| A/G | 79 | 70 | 58 | 21 | NS | NS | NS | ||||
Pc: P-value of Fisher’s exact test with Bonferroni-correction for multiple testing; RR: Relative Risks, CL: 95% Confidence Limits of RR; n: number of chromosomes; N: Number of patients, PcPPV: Prevalence corrected Positive Predictive Value; NS: Not Significant, *= 95%CL for B2AR G allele (1.06-3.29) and for A allele (0.30-0.94)
Table 1: The distribution of alleles and genotype frequencies in 150 Iranian AD patients and 150 healthy controls.
Statistical analysis of TLR2, PICALM (rs3851179), and BDNF alleles and genotypes did not display any significant association with Alzheimer’s disease in total AD, SAD, and FAD patients when compared to controls.
Possible interaction of B2-AR, TLR2, PICALM, and BDNF alleles or genotypes with APOE ε4 allele.
In our preparatory studies, we examined the association of the APOE ε4 allele and AD in a similar patient population [10]. Data from this study and the previously mentioned studies were collected for meta-analysis to achieve more valid conclusions. The distribution of B2-AR, TLR2, PICALM, and BDNF alleles and genotypes frequencies in AD patients and healthy controls, after excluding APOE ε4 genotype, are presented in Table 2. After stratification for the APOE ε4 genotype, it was observed that the frequency of TLR2 and BDNF alleles between APOE ε4-negative AD patients and the controls were not considerably different. While, the frequency of the B2-AR GG homozygote genotype in AD patients who did not carry APOE ε4 allele was significantly increased (Pc=0.008, RR=2.25, 95%CL= 0.33-0.83). Since B2-AR A allele is protective and G allele present susceptibility and on the other hand, B2-AR AA and AG genotypes do not show any significant association with AD while, B2-AR GG homozygote genotype indicates significant association with AD (Pc=0.008, RR=2.25, 95%CL=0.33-0.83) therefore, based on Mendelian inheritance B2-AR G allelic gene is a recessive gene and provide susceptibility in homozygote form for AD.
| All AD vs. controls | |||||
|---|---|---|---|---|---|
| All AD patients | Control | Pc | RR* | PcPPV% | |
| Alleles | |||||
| B2AR | N=134 | N=300 | |||
| A | 33 | 115 | 0.006 | 0.52 | 1.4 |
| G | 101 | 185 | 0.006 | 1.9 | 2.7 |
| TLR2 | N=196 | N=260 | |||
| D | 40 | 60 | NS | ||
| I | 156 | 200 | NS | ||
| PICALM | N=82 | N=260 | |||
| A | 51 | 133 | 0.1 | 1.49 | |
| G | 31 | 127 | 0.1 | 1.04 | |
| BDNF | N=108 | N=196 | |||
| A | 18 | 32 | NS | ||
| G | 90 | 164 | NS | ||
| Genotype | |||||
| B2AR | N=67 | N=150 | |||
| AA | 3 | 18 | 0.12 | 1.22 | |
| AG | 27 | 79 | 0.12 | 1.06 | |
| GG | 37 | 53 | 0.008 | 2.25 | 3.5 |
| TLR2 | N=66 | N=130 | |||
| DD | 2 | 5 | NS | ||
| DI | 27 | 50 | NS | ||
| II | 37 | 75 | NS | ||
| PICALM | N=41 | N=130 | |||
| AA | 15 | 33 | NS | ||
| AG | 21 | 67 | NS | ||
| GG | 5 | 30 | NS | ||
| BDNF | N=54 | N=98 | |||
| AA | 2 | 4 | NS | ||
| AG | 14 | 24 | NS | ||
| GG | 38 | 70 | NS | ||
Pc: P-value of Fisher’s exact test with Bonferroni-correction for multiple testing; RR: Relative Risks; CL 95%: Confidence Limits of RR, n: number of chromosomes; N: Number of patients; NS: Not Significant; PcPPV: Prevalence-corrected Positive Predictive Value; *= 95%CL for B2AR G allele (1.21-3.00), for A allele (0.33-0.83) and GG genotype (1.26-4.06)
Table 2: The distribution of alleles and genotypes frequencies after excluding APOE ε4.
Furthermore, statistical analysis at the allelic level demonstrated a significant association between B2-AR A and G alleles and AD as compared with controls, in that the G allele imposes susceptibility for (Pc=0.006, RR=1.90, 95%CL= 1.21-3.0) and A allele provides protection against AD (Pc=0.006, RR=0.0.52, 95%CL= 0.33-0.83).
Also, after eliminating the effect of APOE ε4, statistical analysis indicated that the frequency of PICALM alleles of A and G in AD patients were increased and decreased, respectively when compared with the controls, but it was not significant after Bonferoni’s correction for multiple testing (Table 1).
Clinical significance of testing for B2-AR A and G alleles and B2-AR GG genotype
The individual absolute risk in the case-control studies can be calculated by PcPPV formula [38]. We employed this formula, which considers the prevalence of AD disease in the general Iranian population, to calculate the PcPPV of testing for B2-AR A and G alleles and B2-AR GG genotype (Table 1). The prevalence of AD for evaluating the predictive values was taken as 2.3% for the Iranian population [39]. The PcPPV of B2-AR A and G alleles were 1.5% and 2.7% respectively in the Iranian FAD patients. Also, the PcPPV of B2-AR GG genotype was the highest among other alleles (PcPPV = 3.5%), after excluding the patients carrying APOE ε4 genotype (Table 2). This indicated that non-carriers of APOE ε4 who carry the B2- AR GG genotype had 3.5% absolute risk of developing AD. These results reinforce the susceptibility role for B2-AR G and the protective role for B2-AR A alleles in the Iranian AD patients.
Response to Rivastigmine
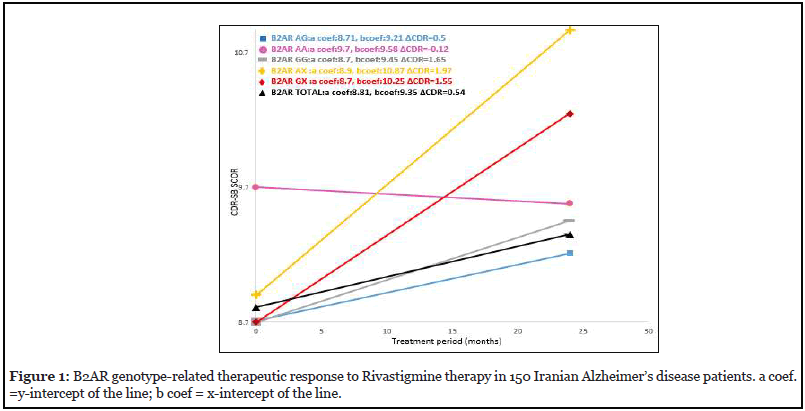
The severity of AD in patients was calculated by CDRSB scale. This calculation was first done at the time of sampling and then after two-year patient follow-up under Rivastigmine treatment. The calculations for the two regimens in this study are signified as CDR1 (initial treatment) and CDR2 (after 24-month treatment), respectively, and ΔCDR=CDR2-CDR1. In this way, the genotype-specific drug response in AD patients was examined for B2-AR, TLR2, PICALM, and BDNF genotypes. Analysis of Genotype-Related Drug Responses to Rivastigmine therapy during 24- month follow-up indicated that patients withB2-AR homozygous GG or heterozygous GA genotypes were the worst responders to drug therapy (ΔCDR=1.65 and 1.97 respectively) when compared to total AD patients (baseline) (ΔCDR=0.54) (Figure 1). Interestingly, patients with B2-AR homozygous AA genotype, had a positive response to the drug and decreasing trend in the disease severity and symptoms (ΔCDR=-0.12) (Figure 1).
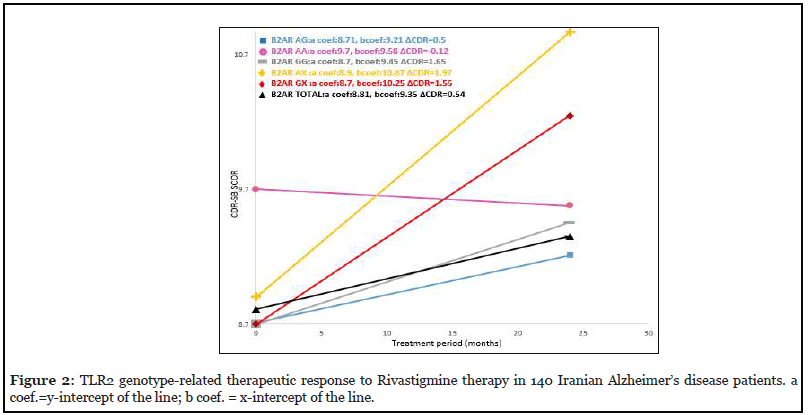
The patients with TLR2 DD genotype exhibited CDR1=5.9 at the start of the drug treatment which was lower than what was calculated for the total AD patients (CDR1=8.81). After 24 months of treatment, these calculated values were ΔCDR2=0.2 for the patients carrying TLR2 DD genotype and ΔCDR2=0.54 for the total AD patients (Figure 2). Note that patients with TLR2 DD genotype have lower ΔCDR2 value than that of the patients with B2-AR AX genotype.
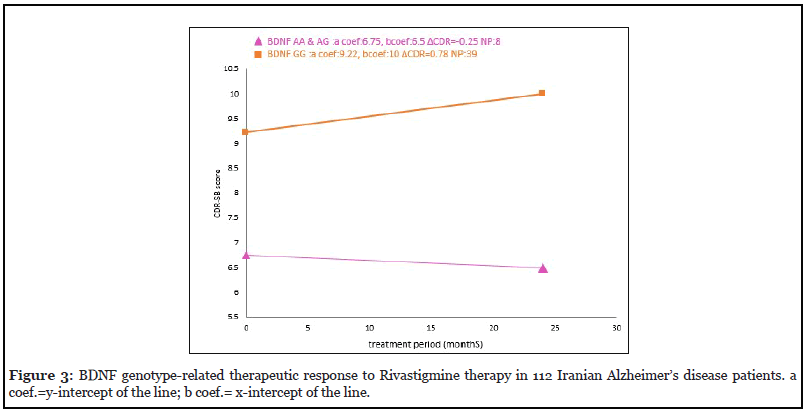
The two-year drug response was also investigated for the patients who were carriers of BDNF genotypes. These results indicated that AD patients with BDNF AX genotype as compared with the total AD patients, exhibited downward trend in the disease severity and symptoms (ΔCDR=-0.25. This finding indicates that AD Patients carrying the BDNF AX genotype present the best response to Rivastigmine therapy (Figure 3).
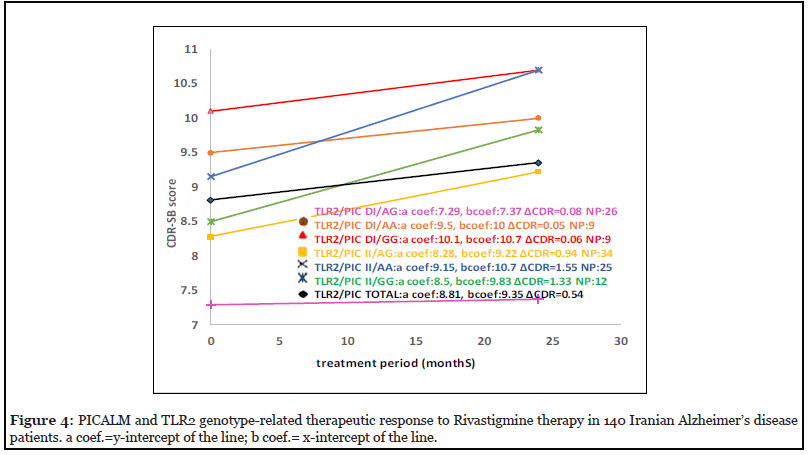
Bigenic genotype interaction in response to Rivastigmine
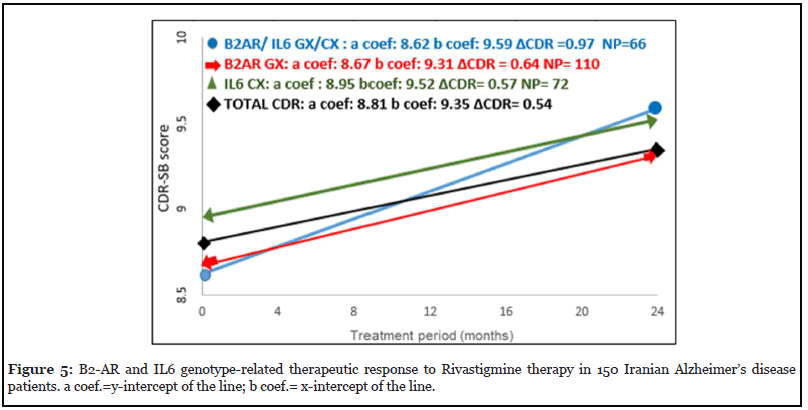
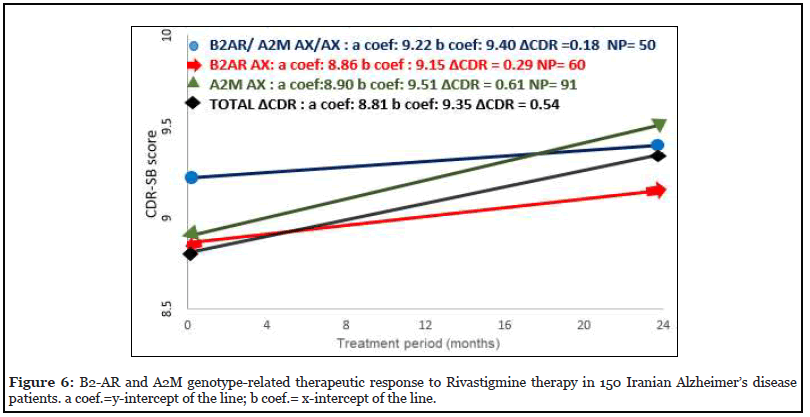
The bigenic genotype-related response of Alzheimer’s patients to Rivastigmine therapy for TLR2 and PICALM genotypes is shown in Figure 4. The calculated ΔCDR during the two-year Rivastigmine follow-up indicated that patients with TLR2 DI-PICALM AG bigenic genotype were good responders (ΔCDR=0.08) and TLR2 II- PICALM AA genotype carriers were bad responders to Rivastigmine (ΔCDR=1.55) (Figure 4). Moreover, we check out interactions between the genes related to this study and the genes studied previously in the same patients for response to Rivastigmine (43). The results of ΔCDR calculation show patients with B2-AR AX- A2M AX bigenic genotype indicated good response to Rivastigmine (ΔCDR=0.18) and B2-AR GX- IL6 GX carriers were bad responder to Rivastigmine treatment (ΔCDR= 0.97) (Figures 5 and 6).
Discussion
Alzheimer’s disease is a complex and multifactorial disease with diverse genetic and environmental contributing factors [1,2]. Many studies indicate that this disease has genetic essence and having a first-degree relative with AD strengthens the risk of developing the disease as much as four times [40]. AD etiology was studied by many researchers who suggested that number of genes contributed to AD risk [5,9,10]. With regard to the complexity of the disease, with numerous possible associated gene mutations, it is highly probable that the underlying genetic predisposition to Alzheimer’s disease could differ among various populations [5-7]. With the purpose of establishing the genetic profile of AD in the Iranian patients, we previously investigated the association of AD with several genes [10,41-43]. In the present study, the association of B2-AR, TLR2, PICALM and BDNF genotypes with the development of AD were examined. Furthermore, the effect of Rivastigmine therapy on Alzheimer’s disease in the Iranian population in B2-AR, TLR2, PICALM and BDNF polymorphic genes was assessed.
Our statistical analyses indicate that between the total AD patients and the control group, there are no significant differences in the frequency of B2-AR alleles. But after stratifying the AD patients into FAD and SAD groups, a significantly positive association of B2-AR G allele (Pc=0.02, RR=1.21, 95%Cl=1.06-3.29) and negative association of allele A (Pc=0.02, RR=0.65, 95%Cl=0.30- 0.94) with FAD was observed. After eliminating the effect of APOE ε4, those patients associated only with APOE ε4 and not associated with second gene are deleted, by eliminating patients carrying APOE ε4 the frequency of second associated gene (e.g. B2-AR GG) is increased and consequently association of second gene becomes more significant. In this study, after adjusting for the effect of APOE ε4, a significantly positive association was identified between B2-AR GG homozygote genotype and AD (Pc=0.008, RR=2.25, 95%Cl= (1.26-4.06). While B2- AR homozygote AA and heterozygote AG genotypes did not demonstrate any significant association with AD, this finding suggest that B2-AR G allelic gene is a recessive gene and predispose to AD in homozygote form of B2-AR GG genotype. Furthermore, association of B2-AR A and G alleles with AD become more significant when APOE ε4 allele is excluded (G allele: Pc=0.006, RR=1.90, 95%Cl= 1.20-3.00, and A allele: Pc=0.006, RR=0.52, 95%Cl= (0.33-083) (Table 2). Based on these results, therefore, we can conclude that the G allele of B2-AR impose predisposition while the A allele confers protection to AD. This finding is in line with the investigation performed in Chinese populations [14]. Increasing evidence has suggested that activation of B2-adrenergic receptors (B2- AR) enhances the gamma-secretase activity and in turn Aβ production. Several single nucleotide polymorphisms (SNPs) have been detected in B2-AR gene; one of them occurring at codons 16 (Gly or Arg). Many studies have observed that this SNP might alter cellular trafficking and receptor desensitization [15-17]. Abnormal activation of B2-AR gene might contribute to Aβ accumulation, one of the hallmarks of AD pathogenesis. Therefore, it can be hypothesized that A allele as a protective effect can enhance, while the G allele as a susceptibility force can reduce the agonist-mediated desensitization of B2-AR, and hence potentially contribute to the pathogenesis of AD [44]. The patients carrying the Gly16 allele of B2-AR (the G allele) have enhanced responsiveness to endogenous norepinephrine which is progressively synthesized by the impact of environmental factors such as stress. Higher levels of norepinephrine provoke increased endocytosis particularly that of presenilin-1(PS1). Subsequently, the increased traffic of PS1 to the late endosomes and lysosomes will provide an optimal environment for γ-secretase activity which leads to more Aβ production. Therefore, individuals with Gly16 allele of B2-AR are more susceptible to AD [45-47].
Growing evidence shows that microglia, which are activated by Amyloid β peptides, play a key role in pathogenesis of AD [48,49]. Chen et al., in 2005 showed that activation of toll-like receptors (TLRs) on microglia promote cellular uptake of Aβ peptides. Also, TLR2 plays a potential role in stimulating the cellular response of microglia to the neurotrophic factors secreted in inflammatory lesions of the neuronal tissue. Interestingly, the results collected here show that none of the TLR2 alleles and genotypes have any impact on the pathogenesis of AD in the Iranian patient population. In contrast to our findings, several studies have shown that TLR2 genotype imposed susceptibility to AD [21,22]. While in other studies and in line with our findings, the authors did not find any association between TLR2 (-196 TO -174 del) polymorphism and AD [50]. Phosphatidylinositol binding Clathrin assembly protein (PICALM) encodes a protein that plays a role in collecting Clathrin. This gene contributes to the pathogenesis of AD through intervening the production, transportation, cleaning, and other pathways related to Aβ [26]. Our observations show no indications of significant differences in infrequencies of PICALM genotypes in total AD patients when compared to controls.
After eliminating the effect of APOE ε4 gene, as compared with controls, the frequency of PICALM allele A and G in the Iranian AD patients was increased and decreased, respectively, but no statistically significant differences were observed. Opposite to this finding however, evidence of association between PICALM GG genotype and increased risk of AD in two studies on Brazilian (P-value=0.007) and Chins (P-value<0.05) patients were reported [51,52]. Interestingly, in another study conducted by Jiang et al. in Han Chinese, the association between the PICALM gene and Alzheimer’s disease was not observed [53]. One of the notable events in the pathogenesis of AD is reduced expression of neurotrophic factors and their receptors, such as Brain-derived neurotrophic factor (BDNF) [27]. This factor promotes neuronal survival and regulates the proliferation and differentiation of nerve cells in the peripheral and central nervous systems. BDNF also facilitates the release of acetylcholine from the end of hippocampal cholinergic neurons [54]. In agreement with the studies of Desai et al. [55] and He et al. [12], our results demonstrated that between the FAD, or SAD patients and controls, there are no significant differences in the frequencies of BDNF genotypes (Table 1 and 2); this observation is contrary to the study presented by Huang et al. [12]. Expectedly, in case-control studies with diverse populations, and small sample sizes, different and sometimes contradictory results were reported. Differences in ethnic origins in addition to the heterogeneity of AD disease may justify the discordance in results obtained from different populations in the world. For instance, Zamani et al. reported a significant difference in frequency and distribution of cystic fibrosis (CF) gene mutations between the population of Iran and those of its neighboring countries such as Pakistan, Turkey, and the Arab countries [56].
The effectiveness of testing for particular allele or genotype can be assessed by employing PcPPV formula [38]. The lifelong prevalence of a disease in a population is needed to measure the PcPPV in that population; the prevalence of AD is 2.3% in the general Iranian population [41]. The PcPPV of B2-AR G allele was 2.7% in FAD patients (Table 1), while after excluding APOE ε4 allele, this number for the homozygous B2-AR GG increased to 3.5% (Table 2). This means that a person who carries the B2-AR GG genotype has a 3.5% absolute risk for developing AD, increasing the risk of AD to 1.5 times higher than what is calculated for the general Iranian population (2.23%). On the other hand, the PcPPV of testing for B2-AR A allele was 1.5% for all FAD patients in the Iranian population (Table 1). This result, therefore, strengthens the notion that B2- AR A allele plays a protective role in developing AD in the Iranian population.
In recent years, numbers of studies have investigated the response of Alzheimer’s patients to Rivastigmine drug, in the context of certain allelic forms of APOE, IL6, A2M and TOMM40 genes. These studies suggested a genotypespecific response to the drug, facilitating a targeted and advantageously economical treatment for AD [10,36]. Interestingly, it has also been reported that only up to 50% of Alzheimer’s patients responded in acceptable levels to acetylcholine esterase inhibitors [57] which is in line with the reported differential allelic effects on such response. Furthermore, research conducted in 2015 indicated that interactions between or independent combinations of different genes could also affect drug responses in AD patients; the presence of certain combined genotypes elicited the best responses, while others elicited the worst responses to Rivastigmine treatment [32]. In this study, we used the CDR-SB scale for determining the effectiveness of Rivastigmine in 140 Iranian Alzheimer’s patients. The results after 24 months of follow-up treatment indicate that, as compared with total patients (ΔCDR=0.54), the patients carrying BDNF AA and AG genotypes had the best response (ΔCDR=-.025), while the patients carrying genotype B2-AR AX had the worst response (ΔCDR=1.97) to Rivastigmine. From this finding, it can be speculated that impaired secretion of BDNF in carriers of AA allele could induce the levels of cholinesterases, hence reduced acetylcholine levels. Therefore, anticholinesterase drugs such as Rivastigmine could bring improvements in the memory and cognitive conditions of such patients. Likewise, considering the role of PICALM protein in endocytosis of acetylcholine in the synaptic regions, the positive response of PICALM AG carriers may have been reinforced by the impact of Rivastigmine. Finally, our data indicate that Alzheimer’s patients with bigenic genotype TLR2 DI-PICALM AG (ΔCDR=0.08) respond positively, while carriers of TLR2 II-PICALM AA combined genotypes respond negatively to Rivastigmine therapy (ΔCDR=1.55). Also, according to the bigenic genotype response to Rivastigmine in relation to interaction between these genes and other genes studied in the same population before, patients with B2-AR AX- A2M AX genotype were the good responder (ΔCDR=0.18) and B2-AR GX- IL6 GX carriers were bad responder to Rivastigmine therapy (ΔCDR= 0.97) (Figures 5 and 6).
These findings suggest that in addition to the effect of single gene polymorphisms, gene-gene interactions or none interacting combination of number of genes might also impact the response of the Alzheimer patients to Rivastigmine therapy.
In Conclusion, our findings suggest that the B2-AR A allele plays a protective role in AD and patients with this allele respond positively to Rivastigmine treatment (ΔCDR=0.24). In contrast, B2-AR G allele imposes susceptibility for AD and the patients carrying this allele exhibited the worst response to Rivastigmine therapy (ΔCDR=0.81). Furthermore, after eliminating the effect of APOE ε4 gene, as compared with controls, PICALM A allele exhibited positive (P=0.05, RR=1.49), while the G allele exhibited negative (P=0.05, RR=1.05) associations with AD in our selected Iranian patients. Also, AD Patients carrying the BDNF AX genotype revealed the best response to Rivastigmine therapy (ΔCDR=-0.25). Studies with larger and more diverse patient populations are required to be determined the exact role of B2-AR, TLR2, PICALM, and BDNF polymorphic genes and their possible contributing factors in Alzheimer’s disease.
Funding
This study was supported by a research grant from Tehran University of Medical Sciences.
Conflicts of Interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Author Contributions
Parvin Mohabattalab: Conceptualization, Methodology, Formal analysis, Writing - original draft, Visualization. Fatemeh Rezaei Rad: Writing - review & editing, Software, Form analysis. Hamid Zamani: Conceptualization, Methodology, Software, Form analysis, Resources, Writing. Fariba Shirvani: Writing - review & editing. Mahdi Zamani: Conceptualization, Methodology, Resources, Investigation, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Compliance with Ethical Standards
This study was certified by the ethics committee and review board of the Tehran University of Medical Sciences.
Research Involving Human Participants and Informed consent
Questionnaire forms were prepared for each patient and control individuals. Before being included in the study, informed consent was taken from all the patients, or their legal guardians and control subjects.
Consent for Publication
All study participants, or their legal guardian, provided informed written consent for publication.
Acknowledgments
We are extremely grateful to the participation of the patients with AD and healthy volunteers in this study.
References
2. Cacabelos R, Alvarez A, Lombardi V, Fernandez-Novoa L, Corzo L, Perez P, et al. Pharmacological treatment of Alzheimer disease: from psychotropic drugs and cholinesterase inhibitors to pharmacogenomics. Drugs Today. 2000 Jul 1;36(7):415-99.
3. Zhang YW, Thompson R, Zhang H, Xu H. APP processing in Alzheimer’s disease. Molecular Brain. 2011 Dec;4(1):1-3.
4. Rocca WA, Hofman A, Brayne C, Breteler MM, Clarke M, Copeland JR, et al. Frequency and distribution of Alzheimer’s disease in Europe: a collaborative study of 1980–1990 prevalence findings. Annals of Neurology: Official Journal of the American Neurological Association and the Child Neurology Society. 1991 Sep;30(3):381-90.
5. Chen SY, Chen TF, Lai LC, Chen JH, Sun Y, Wen LL, et al. Sequence variants of interleukin 6 (IL-6) are significantly associated with a decreased risk of late-onset Alzheimer’s disease. Journal of Neuroinflammation. 2012 Dec;9(1):1-9.
6. Flex A, Giovannini S, Biscetti F, Liperoti R, Spalletta G, Straface G, Landi F, Angelini F, Caltagirone C, Ghirlanda G, Bernabei R. Effect of proinflammatory gene polymorphisms on the risk of Alzheimer’s disease. Neurodegenerative Diseases. 2014;13(4):230-6.
7. Rasmussen L, Delabio R, Horiguchi L, Mizumoto I, Terazaki CR, Mazzotti D, et al. Association between interleukin 6 gene haplotype and Alzheimer’s disease: a Brazilian case-control study. Journal of Alzheimer’s Disease. 2013 Jan 1;36(4):733-8.
8. Behjati F, Atri M, Najmabadi H, Nouri K, Zamani M, Mehdipour P. Prognostic value of chromosome 1 and 8 copy number in invasive ductal breast carcinoma among Iranian women: an interphase FISH analysis. Pathology & Oncology Research. 2005 Sep;11(3):157-63.
9. Edwards TL, Pericak-Vance M, Gilbert JR, Haines JL, Martin ER, Ritchie MD. An association analysis of Alzheimer disease candidate genes detects an ancestral risk haplotype clade in ACE and putative multilocus association between ACE, A2M, and LRRTM3. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2009 Jul 5;150(5):721-35.
10. Zamani M, Mehri M, Kollaee A, Yenki P, Ghaffarpor M, Harirchian MH, et al. Pharmacogenetic study on the effect of rivastigmine on PS2 and APOE genes in Iranian Alzheimer patients. Dementia and Geriatric Cognitive Disorders extra. 2011;1(1):180-9.
11. Carrasquillo MM, Belbin O, Hunter TA, Ma L, Bisceglio GD, Zou F, et al. Replication of CLU, CR1, and PICALM associations with alzheimer disease. Archives of Neurology. 2010 Aug 1;67(8):961-4.
12. He XM, Zhang ZX, Zhang JW, Zhou YT, Tang MN, Wu CB, et al. Lack of association between the BDNF gene Val66Met polymorphism and Alzheimer disease in a Chinese Han population. Neuropsychobiology. 2007;55(3- 4):151-5.
13. Kim SH, Mun MJ, Kim JH, Jang WC. Associations of Three Polymorphisms in Endocytosis-related Genes with the Risk of Alzheimer’s Disease in Korean and East Asian Populations. of. 2016;7:2.
14. Yu JT, Tan L, Ou JR, Zhu JX, Liu K, Song JH, et al. Polymorphisms at the β2-adrenergic receptor gene influence Alzheimer’s disease susceptibility. Brain Research. 2008 May 19;1210:216-22.
15. Green SA, Liggett SB. A proline-rich region of the third intracellular loop imparts phenotypic beta 1-versus beta 2-adrenergic receptor coupling and sequestration. Journal of Biological Chemistry. 1994 Oct 21;269(42):26215-9.
16. Green SA, Turki J, Bejarano P, Hall IP, Liggett SB. Influence of beta 2-adrenergic receptor genotypes on signal transduction in human airway smooth muscle cells. American Journal of Respiratory Cell and Molecular Biology. 1995 Jul;13(1):25-33.
17. Moore PE, Laporte JD, Abraham JH, Schwartzman IN, Yandava CN, Silverman ES, et al. Polymorphism of the ?2-adrenergic receptor gene and desensitization in human airway smooth muscle. American Journal of Respiratory and Critical Care Medicine. 2000 Dec 1;162(6):2117-24.
18. Igbavboa U, Johnson-Anuna LN, Rossello X, Butterick TA, Sun GY, Wood WG. Amyloid beta-protein1-42 increases cAMP and apolipoprotein E levels which are inhibited by ?1 and ?2-adrenergic receptor antagonists in mouse primary astrocytes. Neuroscience. 2006 Oct 27;142(3):655-60.
19. Lee RK, Araki W, Wurtman RJ. Stimulation of amyloid precursor protein synthesis by adrenergic receptors coupled to cAMP formation. Proceedings of the National Academy of Sciences. 1997 May 13;94(10):5422-6.
20. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009 Oct;41(10):1088-93.
21. Sohrabifar N, Gharesouran J, Talebi M, Ghojazadeh M, Ardebili SM. Association of CLU and TLR2 gene polymorphisms with late-onsetAlzheimer disease in a northwestern Iranian population. Turkish Journal of Medical Sciences. 2015 Oct 26;45(5):1082-6.
22. Yu JT, Mou SM, Wang LZ, Mao CX, Tan L. Toll- like receptor 2-196 to-174 del polymorphism influences the susceptibility of Han Chinese people to Alzheimer’s disease. Journal of Neuroinflammation. 2011 Dec;8(1):1-4.
23. Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nature Genetics. 2009 Oct;41(10):1088-93.
24. Santos-Rebouças CB, Gonçalves AP, Dos Santos JM, Abdala BB, Motta LB, Laks J, de Borges MB, de Rosso AL, Pereira JS, Nicaretta DH, Pimentel MM. rs3851179 polymorphism at 5′ to the PICALM gene is associated with Alzheimer and Parkinson diseases in Brazilian population. Neuromolecular Medicine. 2017 Sep;19(2):293-9.
25. Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA, et al. Age at onset in two common neurodegenerative diseases is genetically controlled. The American Journal of Human Genetics. 2002 Apr 1;70(4):985-93.
26. Xu W, Tan L, Yu JT. The role of PICALM in Alzheimer’s disease. Molecular Neurobiology. 2015 Aug;52(1):399-413.
27. Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Research Reviews. 2008 Nov 1;59(1):201-20.
28. Baj G, Carlino D, Gardossi L, Tongiorgi E. Toward a unified biological hypothesis for the BDNF Val66Metassociated memory deficits in humans: a model of impaired dendritic mRNA trafficking. Frontiers in Neuroscience. 2013 Oct 30;7:188.
29. Egan MF, Kojima M, Callicott JH, Goldberg TE, Kolachana BS, Bertolino A, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003 Jan 24;112(2):257-69.
30. Cacabelos, R.J.D.N. and Perspectives, Pharmacogenomics in Alzheimer’s Disease. 2000. 13(4): p. 252-254.
31. Spencer CM, Noble S. Rivastigmine. Drugs & aging. 1998 Nov;13(5):391-411.
32. Cacabelos R, Cacabelos P, Torrellas C, Tellado I, Carril JC. Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Pharmacogenomics in Drug Discovery and Development. 2014:323-556.
33. Braun CM, Daigneault R, Gaudelet S, Guimond A. Diagnostic and Statistical Manual of Mental Disorders, symptoms of mania: which one (s) result (s) more often from right than left hemisphere lesions?. Comprehensive Psychiatry. 2008 Sep 1;49(5):441-59.
34. Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975 Nov 1;12(3):189-98.
35. Kim SH, Oh SY, Oh HB, Kim YK, Cho SH, Kim YY, et al. Association of β2-adrenoreceptor polymorphisms with nocturnal cough among atopic subjects but not with atopy and nonspecific bronchial hyperresponsiveness. Journal of Allergy and Clinical Immunology. 2002 Apr 1;109(4):630- 5.
36. Gharesouran J, Rezazadeh M, Khorrami A, Ghojazadeh M, Talebi M. Genetic evidence for the involvement of variants at APOE, BIN1, CR1, and PICALM loci in risk of late-onset Alzheimer’s disease and evaluation for interactions with APOE genotypes. Journal of Molecular Neuroscience. 2014 Dec;54(4):780-6.
37. Borroni B, Grassi M, Archetti S, Costanzi C, Bianchi M, Caimi L, et al. BDNF genetic variations increase the risk of Alzheimer’s disease-related depression. Journal of Alzheimer’s Disease. 2009 Jan 1;18(4):867-75.
38. Zamani M, Cassiman JJ. Reevaluation of the importance of polymorphic HLA class II alleles and amino acids in the susceptibility of individuals of different populations to type I diabetes. American Journal of Medical Genetics. 1998 Mar 5;76(2):183-94.
39. Gholamzadeh S, Heshmati B, Mani A, Petramfar P, Baghery Z. The prevalence of alzheimer’s disease; its risk and protective factors among the elderly population in Iran. Shiraz E-Medical Journal. 2017 Sep 30;18(9).
40. Abraham R, Moskvina V, Sims R, Hollingworth P, Morgan A, Georgieva L, et al. A genome-wide association study for late-onset Alzheimer’s disease using DNA pooling. BMC Medical Genomics. 2008 Dec;1(1):1-3.
41. Rad FR, Akbari MG, Zamani M, Bayat S, Zamani M. Pharmacogenetic and Association Studies on the Influence of HLA Alleles and Rivastigmine on the Iranian Patients with Late-Onset Alzheimer’s Disease. Molecular Neurobiology. 2021 Jun;58(6):2792-802.
42. Sayad A, Noruzinia M, Zamani M, Harirchian MH, Kazemnejad A. Association study of cathepsin D gene polymorphism in Iranian patients with sporadic late-onset Alzheimer’s disease. Dementia and Geriatric Cognitive Disorders. 2014;37(5-6):257-64.
43. Zamani M, Mohammadi M, Zamani H, Tavasoli A. Pharmacogenetic Study on the Impact of Rivastigmine Concerning Genetic Variants of A2M and IL-6 Genes on Iranian Alzheimer’s Patients. Molecular Neurobiology. 2016 Sep;53(7):4521-8.
44. Dishy V, Sofowora GG, Xie HG, Kim RB, Byrne DW, Stein CM, et al. The effect of common polymorphisms of the β2-adrenergic receptor on agonist-mediated vascular desensitization. New England Journal of Medicine. 2001 Oct 4;345(14):1030-5.
45. Langui D, Girardot N, El Hachimi KH, Allinquant B, Blanchard V, Pradier L, et al. Subcellular topography of neuronal A? peptide in APPxPS1 transgenic mice. The American Journal of Pathology. 2004 Nov 1;165(5):1465-77.
46. Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, et al. Oligomerization of Alzheimer’s β-amyloid within processes and synapses of cultured neurons and brain. Journal of Neuroscience. 2004 Apr 7;24(14):3592-9.
47. Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, et al. Association of γ-secretase with lipid rafts in post- Golgi and endosome membranes. Journal of Biological Chemistry. 2004 Oct 22;279(43):44945-54.
48. Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiology of Aging. 2000 May 1;21(3):383-421.
49. Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response?. Nature Medicine. 2006 Sep;12(9):1005-15.
50. Yu JT, Sun YP, Ou JR, Cui WZ, Zhang W, Tan L. No association of toll-like receptor 2 polymorphisms with Alzheimer’s disease in Han Chinese. Neurobiology of Aging. 2011 Oct 1;32(10):1924-e1.
51. Belcavello L, Camporez D, Almeida LD, Morelato RL, Batitucci MC, de Paula F. Association of MTHFR and PICALM polymorphisms with Alzheimer’s disease. Molecular Biology reports. 2015 Mar 1;42(3):611-6.
52. Sun DM, Chen HF, Zuo QL, Su F, Bai F, Liu CF. Effect of PICALM rs3851179 polymorphism on the default mode network function in mild cognitive impairment. Behavioural Brain Research. 2017 Jul 28;331:225-32.
53. Jiang T, Yu JT, Tan MS, Wang HF, Wang YL, Zhu XC, et al. Genetic variation in PICALM and Alzheimer’s disease risk in Han Chinese. Neurobiology of Aging. 2014 Apr 1;35(4):934-e1.
54. Knipper M, da Penha Berzaghi M, Blöchl A, Breer H, Thoenen H, Lindholm D. Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. European Journal of Neuroscience. 1994 Apr;6(4):668-71.
55. Desai P, Nebes R, DeKosky ST, Kamboh MI. Investigation of the effect of brain-derived neurotrophic factor (BDNF) polymorphisms on the risk of late-onset Alzheimer’s disease (AD) and quantitative measures of AD progression. Neuroscience Letters. 2005 May 13;379(3):229-34.
56. Alibakhshi R, Kianishirazi R, Cassiman JJ, Zamani M, Cuppens H. Analysis of the CFTR gene in Iranian cystic fibrosis patients: identification of eight novel mutations. Journal of Cystic Fibrosis. 2008 Mar 1;7(2):102-9.
57. Schneider LS, Farlow MR. Predicting response to cholinesterase inhibitors in Alzheimer’s disease. CNS Drugs. 1995 Aug;4(2):114-24.