Keywords
Estrogen, Estrogen receptor alpha, Estrogen receptor beta, Inflammatory bowel disease, Crohn’s disease, Inflammation, SAMP ileitis
Introduction
Inflammatory bowel diseases (IBD) are inflammatory conditions characterized by chronic and recurrent intestinal inflammation [1,2]. IBD affects nearly five million people worldwide, with an estimated prevalence of 245.3 cases per 100,000 people in the United States alone [3]. The incidence of IBD is steadily increasing in industrialized countries and is appearing more often developing countries, contributing to its rise as a global disease burden [4,5].
IBD encompasses ulcerative colitis (UC), in which ulcers and inflammatory patches are localized to the innermost lining of the colon [1], and Crohn’s disease (CD), which has discontinuous patches of inflammation anywhere in the gastrointestinal tract [1]. IBD patients experience symptoms that range in severity, including fatigue, weight loss, blood in stool, and abdominal pain [1]. These symptoms can be debilitating and lead to life-threatening complications, thus impeding quality of life. Symptoms of IBD are observed at all ages, including in childhood. Pediatric IBD patients experience impaired growth development, primarily seen as reduced height and weight measurements compared to healthy peers [6,7]. Nearly 1 out of 4 IBD patients are diagnosed before 18 years of age, and one quarter of affected adolescents are under 10 years of age when diagnosed [8].
Although the exact etiology of IBD is not conclusive, there are numerous factors that are believed to be involved, including genetic predisposition, environmental exposure, and lifestyle [9,10]. Combinations of these factors can lead to physiological changes causing dysregulation of immune responses, mucosal barrier function, and gastrointestinal microbiota composition [11]. Because the causes driving IBD are not fully understood and there is no known cure for IBD, patients turn to costly treatments for managing symptoms. These treatments result in healthcare costs of nearly $23,000 per year [12], with costs driven by treatment of comorbidities (ie. anemia, mental health), emergency room visits, and pharmacotherapies [12,13]. Many treatments use pharmacological intervention to modulate immune responses, primarily by reducing lymphocyte effector function, reducing circulating levels of pro-inflammatory cytokines, and promoting regulatory T cell (Treg) function [14].
17β-estradiol (estrogen, E2) regulates numerous cellular processes, including proliferation, angiogenesis, and inhibition of apoptosis [15]. E2 generally has an immunoprotective role, in which E2 signaling promotes pathogen clearance and tissue repair [16], in part by promoting differentiation of anti-inflammatory immune cell populations including M2 macrophages [17], Th2 cells [18], and Tregs [19]. E2 signals through the estrogen receptors alpha and beta (ERα and ERβ), which are part of the nuclear receptor family [20]. Upon ligand binding, ERs either homo- or hetero-dimerize and translocate to the nucleus, where they modulate transcriptional changes in target genes [21,22]. ERs can bind directly or indirectly (via recruitment of transcriptional coactivators) to estrogen response elements (EREs) on promoter regions of target genes [23,24]. ERα is known to have more pro-inflammatory and pro-proliferative roles, whereas ERβ has been shown to confer a protective role that limits inflammation [25,26].
Dysregulated E2 signaling contributes to progression of inflammatory conditions, such as IBD [14,27]. Patients with active IBD (both UC and CD) have reduced ERβ expression in inflamed colonic mucosal tissues [28–30]. Additionally, previous work from our lab demonstrated that peripheral Tregs from female patients with active CD have significantly reduced expression of ERβ [26]. Together, these suggest that ERβ is important for regulating inflammation in the intestine, and that the normally protective role of ERβ as an “inflammatory brake” is impaired in IBD, especially in female patients. This aligns with other clinical observations of IBD, including that disease incidence of CD is higher in adult women than men and that women are more likely to experience more severe disease manifestations [31]. Several reports have suggested that hormones are likely to play a role in the development of disease [32,33], making E2 a promising target for investigating IBD.
Various animal experimental models have been generated to investigate the mechanisms driving intestinal inflammation. Experimental models of IBD have been used to characterize defects in epithelial barrier integrity, innate immune responses, and adaptive immune responses [34]. The SAMP/YitFC (“SAMP”) mouse model is one such model, which arose spontaneously from a strain of senescence accelerated mice [35] and exhibit Crohn’s-like ileitis that mimics the pathology and female sex bias of human CD [36]. Female SAMP mice display earlier onset and increased severity of ileitis compared to male SAMP [27]. Acute and chronic inflammation are localized to the terminal ileum, where inflammation is transmural and has a discontinuous pattern [36]. Disruption of the epithelial barrier is present, along with crypt elongation and tissue atrophy [37]. Histological analyses show infiltration of various cell types, including lymphocytes, neutrophils, and macrophages [37]. SAMP mice exhibit high IFNγ production by 4 weeks of age [38] and full onset of ileitis by 10 weeks of age [39]. Inflammation progressively worsens as mice age, and by 40-50 weeks they display thickening of the bowel wall and stricture formation in the intestine [39]. While the SAMP model has advantages for modeling chronic and spontaneous IBD, the mechanisms driving disease remain to be fully understood.
Our previous work identified a mechanism by which E2 modulates Treg differentiation and function in SAMP mice. Female SAMP mice with global deletion of ERβ (SAMPΔERβ) exhibited decreased Foxp3 expression in CD4+ T cells from the mesenteric lymph node (mLN), along with altered expression of canonical Treg-associated genes from full thickness ileal tissues, and decreased Treg differentiation and suppressive function [26]. These results were observed in female mice but not males, indicating that deletion of ERβ–thus promoting ERα-specific signaling–has a more profound effect in driving intestinal inflammation in females. Here, we assessed whether ERα would also display sex-specific effects in chronic intestinal inflammation using a similar experimental SAMP model. We hypothesized that eliminating ERα-specific signaling, which is well-known for its pro-inflammatory roles, would reduce the severity of ileitis.
Materials and Methods
Heterozygous ERα+/- mice were back-crossed with SAMP/YitFc mice for eight generations to generate SAMP mice heterozygous for ERα. These heterozygous mice were then bred to generate SAMP mice lacking global expression of ERα (SAMP-ERα-KO), similar to our previously described work in generating SAMPΔERβ mice [26]. Heterozygous mice were used for breeding because mice with ablation of ERα have impaired fertility [40]. Deletion of ERα was confirmed by Western blot. Male and female SAMP-ERα-KO mice were euthanized at 6, 10, 15, and 20 weeks of age and assessed for ileal inflammation. Total inflammatory scores (TIS) were assigned to each mouse based on a set of metrics determined by histological assessment of the degree of ulceration, re-epithelialization, active inflammation, chronic inflammation, and transmural inflammation, calculated by a pathologist blinded to mouse genotype and sex, as previously described [27].
Results
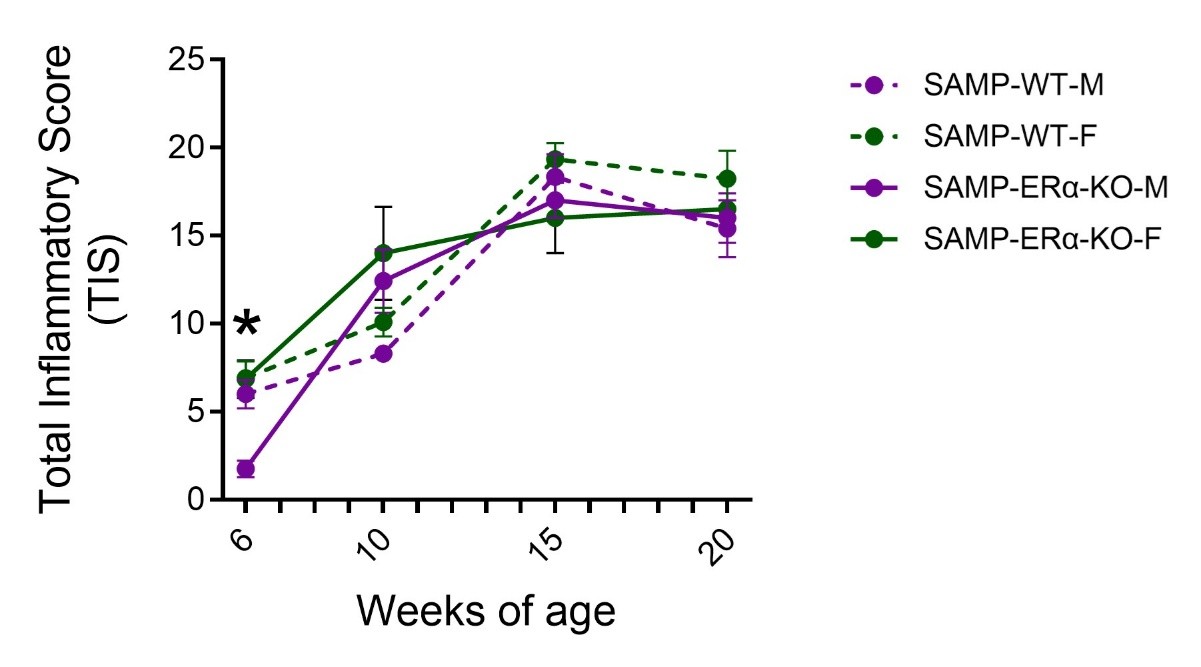
To assess the role of ERα on progression of inflammation, ileal tissues were collected from male and female SAMP-ERα-KO mice and evaluated for inflammation by histological staining (H&E). At six weeks of age, female SAMP-ERα-KO mice were found to have significantly higher Total Inflammatory Scores (TIS) compared to males, indicating more severe ileitis (Figure 1). This data complements our previous findings that female native SAMP mice display earlier onset of ileitis compared to males beginning at 6 weeks of age [27]. TIS in native SAMP and SAMP-ERα-KO mice increase dramatically from ages 6 to 10 weeks, but then plateau from ages 10-20 weeks for both males and females [27] (Figure 1). After 10 weeks of age, there are no significant differences in TIS between males and females, demonstrating the loss of female-biased ileitis normally observed in SAMP mice [27]. This suggests that ERα might be necessary to promote ileal inflammation, specifically in females.
Discussion
IBDs exhibit chronic inflammation in the intestine with high rates of relapse. Although the exact mechanisms causing IBD remain to be elucidated, many reports have suggested that E2 signaling is a strong contributing factor. Our previous work showed that female, but not male, SAMP mice with deletion of ERβ had exacerbated symptoms of intestinal inflammation, suggesting that inflammation is driven by ERα-specific signaling [26]. In agreement with our previous work, the current results demonstrate that deletion of ERα results in a similar degree of inflammation between male and female SAMP mice as inflammation becomes more chronic over time (>6 weeks of age). Indeed, the deletion of ERα eliminates the sex bias of exacerbated inflammation normally present in female SAMP mice. These results also suggest that deletion of ERα–thereby promoting ERβ-specific signaling–may be useful in revealing protective roles of ERβ in regulating SAMP ileitis. This complements our previous findings, in which we found that deletion of ERα is protective in chemically-induced colitis [41].
We hypothesized that deletion of ERα would have a protective effect in SAMP mice by reducing overall severity of inflammation. However, our observations were not entirely as expected. SAMP-ERα-KO mice maintained high TIS that increased with age, similar to development of disease in native SAMP [36], demonstrating that eliminating ERα does not prevent inflammation from occurring. However, the degree of inflammation became similar in males and females (Figure 1). TIS from SAMP-ERα-KO mice showed a female sex bias at 6 weeks of age, around the time of puberty [42]. In native SAMP mice, females show earlier onset of inflammation beginning around 6 weeks and experience more severe inflammation throughout the course of disease [27]. This suggests that disease severity worsens when E2 has increased biological activity and more active signaling. This aligns with other clinical findings where women with autoimmune conditions experience enhanced inflammation during puberty, menstruation, or pregnancy, when E2 levels are higher [43]. Additionally, women receiving hormone treatments such as oral contraceptive pills, which raise serum E2 levels, are correlated with increased risk for IBD and worsened symptoms [44,45]. Because we observe in our model that inflammation is similar in males and females beginning at 10 weeks, we can speculate that the lack of ERα-specific signaling may prevent inflammation from getting worse. Collectively, these results suggest a pathogenic role of ERα in driving inflammation in SAMP mice.
Given the known pro-inflammatory roles of ERα, together with higher levels of circulating E2 in women, we can speculate that it has a more pronounced role in enhancing intestinal inflammation in females than males. We can also speculate that a major role of ERβ is to regulate activity of ERα to modulate inflammation. ERβ is the predominant estrogen receptor expressed in the colon and is highly expressed in the intestine compared to other peripheral tissues [46]. ERβ regulates growth, organization, and maintenance of epithelial structure to protect intestinal epithelial barrier integrity and function [47,48]. ERβ also promotes autophagy, an important process in maintaining intestinal homeostasis, by supporting function of intestinal epithelial cells, regulating microbiota, and modulating immune responses by promoting an anti-inflammatory environment [49]. Autophagy is dysregulated in both IBD patients and experimental IBD, and treatment with autophagy agonists has been shown to improve intestinal inflammation [50].
Significant reduction of ERβ expression has been observed in both experimental IBD and in IBD patients [46,51]. Interestingly, decreased ERβ expression is only observed in patients with active IBD, as levels of ERβ are unchanged for individuals in remission [28]. This suggests that relative lack of ERβ signaling (or enhanced ERα signaling) is associated with active inflammation. Restoring ERβ expression and promoting ERβ-specific signaling has been shown to alleviate DSS-induced colitis in mice [29]. Specifically, activation of ERβ resulted in reduced weight loss, lower levels of pro-inflammatory cytokines, and reduced histopathological damage [29]. It is possible that the SAMP-ERα-KO disease model used for our study exhibits improved intestinal epithelial integrity contributing to less severe inflammation in females, but further investigation is needed to better elucidate the mechanisms involved.
Our findings show that diminished ERα-specific signaling (which leads to enhanced ERβ-specific signaling) is beneficial for reducing inflammation in IBD. It is likely in IBD that ERα signaling is unchecked by ERβ, and this lack of regulation enables ERα to promote proinflammatory, mitogenic signaling pathways. Since ERβ expression is reduced in IBD, it could be beneficial for therapies to focus on raising endogenous ERβ expression in the intestine or promoting ERβ-specific signaling. ERβ has been shown to reduce collagen deposition via the TGFβ and TLR4 signaling pathways, therefore alleviating intestinal fibrosis [30], thus making it an attractive target for anti-inflammatory treatment. Additionally, IBD has T cell-driven components that may be mediated by E2 signaling activity. Studies have reported that high levels of E2 induce T cell differentiation and that these generated T cells are thought to be more autoreactive and contribute to higher incidence of autoimmune disease in females [52]. Males, on the other hand, have higher frequencies of Tregs than females, in part due to elevated androgens and decreased ERα signaling [53]. Because estrogen levels are different in males and females, it is important to carefully devise therapies that can promote recovery without many off-target effects. More research is needed to better understand the nuances of ERα- versus ERβ-specific signaling and their roles in inflammatory processes.
Abbreviations
IBD: Inflammatory Bowel Disease; UC: Ulcerative Colitis; CD: Crohn’s Disease; E2: 17β-estradiol, Estrogen; Erα: Estrogen Receptor Alpha; Erβ: Estrogen Receptor Beta; Treg: Regulatory T cell; SAMP: SAMP/YitFC
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This work was supported by the National Institutes of Health (R01DK128143 to WAG, and T32AI089474 to AVB) and the Crohn’s and Colitis Foundation (Senior Research Award #1169305 to WAG).
Author Contributions Statement
AVB: writing – original draft and editing, formal analysis; WAG: writing – review and editing, conceptualization, investigation, funding acquisition, project administration.
References
2. Wallace KL, Zheng LB, Kanazawa Y, Shih DQ. Immunopathology of inflammatory bowel disease. World J Gastroenterol. 2014 Jan 7;20(1):6–21.
3. Wang R, Li Z, Liu S, Zhang D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: a systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open. 2023 Mar 28;13(3):e065186.
4. Kaplan GG. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol. 2015 Dec;12(12):720–7.
5. Molodecky NA, Soon IS, Rabi DM, Ghali WA, Ferris M, Chernoff G, et al. Increasing incidence and prevalence of the inflammatory bowel diseases with time, based on systematic review. Gastroenterology. 2012 Jan;142(1):46–54.e42; quiz e30.
6. Zhou X, Kern I, Rothe U, Schoffer O, Weidner J, Richter T, et al. Growth development of children and adolescents with inflammatory bowel disease in the period 2000-2014 based on data of the Saxon pediatric IBD registry: a population-based study. BMC Gastroenterol. 2024 Jan 9;24(1):25.
7. Ishige T. Growth failure in pediatric onset inflammatory bowel disease: mechanisms, epidemiology, and management. Transl Pediatr. 2019 Jan;8(1):16–22.
8. Benchimol EI, Fortinsky KJ, Gozdyra P, Van den Heuvel M, Van Limbergen J, Griffiths AM. Epidemiology of pediatric inflammatory bowel disease: a systematic review of international trends. Inflamm Bowel Dis. 2011 Jan;17(1):423–39.
9. Ocansey DKW, Zhang L, Wang Y, Yan Y, Qian H, Zhang X, et al. Exosome-mediated effects and applications in inflammatory bowel disease. Biol Rev Camb Philos Soc. 2020 Oct;95(5):1287–307.
10. Ungaro R, Mehandru S, Allen PB, Peyrin-Biroulet L, Colombel JF. Ulcerative colitis. Lancet. 2017 Apr 29;389(10080):1756–70.
11. Ocansey DKW, Qiu W, Wang J, Yan Y, Qian H, Zhang X, et al. The Achievements and Challenges of Mesenchymal Stem Cell-Based Therapy in Inflammatory Bowel Disease and Its Associated Colorectal Cancer. Stem Cells Int. 2020 Mar 18;2020:7819824.
12. Park KT, Ehrlich OG, Allen JI, Meadows P, Szigethy EM, Henrichsen K, et al. The Cost of Inflammatory Bowel Disease: An Initiative From the Crohn's & Colitis Foundation. Inflamm Bowel Dis. 2020 Jan 1;26(1):1–10.
13. Limsrivilai J, Stidham RW, Govani SM, Waljee AK, Huang W, Higgins PD. Factors That Predict High Health Care Utilization and Costs for Patients With Inflammatory Bowel Diseases. Clin Gastroenterol Hepatol. 2017 Mar;15(3):385–92.e2.
14. Cai Z, Wang S, Li J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front Med (Lausanne). 2021 Dec 20;8:765474.
15. Snoj N, Dinh P, Bedard P, Sotiriou C. Molecular Biology of Breast Cancer. In: Coleman WB, Tsongalis GJ, editors. Molecular Pathology. San Diego: Academic Press; 2009. p. 501-17.
16. Briggs J, Teyssier N, Nankabirwa JI, Rek J, Jagannathan P, Arinaitwe E, et al. Sex-based differences in clearance of chronic Plasmodium falciparum infection. Elife. 2020 Oct 27;9:e59872.
17. Keselman A, Fang X, White PB, Heller NM. Estrogen Signaling Contributes to Sex Differences in Macrophage Polarization during Asthma. J Immunol. 2017 Sep 1;199(5):1573–83.
18. Salem ML. Estrogen, a double-edged sword: modulation of TH1- and TH2-mediated inflammations by differential regulation of TH1/TH2 cytokine production. Curr Drug Targets Inflamm Allergy. 2004 Mar;3(1):97–104.
19. Polanczyk MJ, Hopke C, Huan J, Vandenbark AA, Offner H. Enhanced FoxP3 expression and Treg cell function in pregnant and estrogen-treated mice. J Neuroimmunol. 2005 Dec 30;170(1-2):85–92.
20. De Abreu FB, Wells WA, Tsongalis GJ. The emerging role of the molecular diagnostics laboratory in breast cancer personalized medicine. Am J Pathol. 2013 Oct;183(4):1075–83.
21. Parker MG. Transcriptional activation by oestrogen receptors. Biochem Soc Symp. 1998;63:45–50.
22. Fuentes N, Silveyra P. Estrogen receptor signaling mechanisms. Adv Protein Chem Struct Biol. 2019;116:135–70.
23. Hall JM, McDonnell DP, Korach KS. Allosteric regulation of estrogen receptor structure, function, and coactivator recruitment by different estrogen response elements. Mol Endocrinol. 2002 Mar;16(3):469–86.
24. Harding AT, Heaton NS. The Impact of Estrogens and Their Receptors on Immunity and Inflammation during Infection. Cancers (Basel). 2022 Feb 12;14(4):909.
25. Mohammad I, Starskaia I, Nagy T, Guo J, Yatkin E, Väänänen K, et al. Estrogen receptor α contributes to T cell-mediated autoimmune inflammation by promoting T cell activation and proliferation. Sci Signal. 2018 Apr 17;11(526):eaap9415.
26. Goodman WA, Bedoyan SM, Havran HL, Richardson B, Cameron MJ, Pizarro TT. Impaired estrogen signaling underlies regulatory T cell loss-of-function in the chronically inflamed intestine. Proc Natl Acad Sci U S A. 2020 Jul 21;117(29):17166–76.
27. Goodman WA, Garg RR, Reuter BK, Mattioli B, Rissman EF, Pizarro TT. Loss of estrogen-mediated immunoprotection underlies female gender bias in experimental Crohn's-like ileitis. Mucosal Immunol. 2014 Sep;7(5):1255–65.
28. Pierdominici M, Maselli A, Varano B, Barbati C, Cesaro P, Spada C, et al. Linking estrogen receptor β expression with inflammatory bowel disease activity. Oncotarget. 2015 Dec 1;6(38):40443–51.
29. Li J, Chen Y, Yu Q, Li S, Zhang X, Cheng Y, et al. Estrogen receptor β alleviates colitis in intestinal epithelial cells and activates HIF-1a and ATG-9a-mediated autophagy. Exp Cell Res. 2025 Apr 15;447(2):114520.
30. Ling F, Chen Y, Li J, Xu M, Song G, Tu L, et. Estrogen Receptor β Activation Mitigates Colitis-associated Intestinal Fibrosis via Inhibition of TGF-β/Smad and TLR4/MyD88/NF-κB Signaling Pathways. Inflamm Bowel Dis. 2025 Jan 6;31(1):11–27.
31. Saibeni S, Cortinovis I, Beretta L, Tatarella M, Ferraris L, Rondonotti E, et al. Gender and disease activity influence health-related quality of life in inflammatory bowel diseases. Hepatogastroenterology. 2005 Mar-Apr;52(62):509–15.
32. Loftus EV Jr. Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences. Gastroenterology. 2004 May;126(6):1504–17.
33. Rolston VS, Boroujerdi L, Long MD, McGovern DPB, Chen W, Martin CF, et al. The Influence of Hormonal Fluctuation on Inflammatory Bowel Disease Symptom Severity-A Cross-Sectional Cohort Study. Inflamm Bowel Dis. 2018 Jan 18;24(2):387–93.
34. Valatas V, Bamias G, Kolios G. Experimental colitis models: Insights into the pathogenesis of inflammatory bowel disease and translational issues. Eur J Pharmacol. 2015 Jul 15;759:253–64.
35. Takeda T, Hosokawa M, Takeshita S, Irino M, Higuchi K, Matsushita T, et al. A new murine model of accelerated senescence. Mech Ageing Dev. 1981 Oct;17(2):183–94.
36. Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, Mercado JR, et al. SAMP1/YitFc mouse strain: a spontaneous model of Crohn's disease-like ileitis. Inflamm Bowel Dis. 2011 Dec;17(12):2566–84.
37. Matsumoto S, Okabe Y, Setoyama H, Takayama K, Ohtsuka J, Funahashi H, et al. Inflammatory bowel disease-like enteritis and caecitis in a senescence accelerated mouse P1/Yit strain. Gut. 1998 Jul;43(1):71–8.
38. Kosiewicz MM, Nast CC, Krishnan A, Rivera-Nieves J, Moskaluk CA, Matsumoto S, et al. Th1-type responses mediate spontaneous ileitis in a novel murine model of Crohn's disease. J Clin Invest. 2001 Mar;107(6):695–702.
39. Rivera-Nieves J, Bamias G, Vidrich A, Marini M, Pizarro TT, McDuffie MJ, et al. Emergence of perianal fistulizing disease in the SAMP1/YitFc mouse, a spontaneous model of chronic ileitis. Gastroenterology. 2003 Apr;124(4):972–82.
40. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993 Dec 1;90(23):11162–6.
41. Pereda GA, Kocinski AD, Broncano AV, McNeer SK, Raymond ML, Ziats NP, et al. Sex Differences in Colonic Inflammation are Driven by Epithelial-Specific Expression of Estrogen Receptor Alpha. Gastro Hep Adv. 2025 Jan 27;4(5):100624.
42. Mayer C, Acosta-Martinez M, Dubois SL, Wolfe A, Radovick S, Boehm U, et al. Timing and completion of puberty in female mice depend on estrogen receptor alpha-signaling in kisspeptin neurons. Proc Natl Acad Sci U S A. 2010 Dec 28;107(52):22693–8.
43. Mogadam M, Korelitz BI, Ahmed SW, Dobbins WO 3rd, Baiocco PJ. The course of inflammatory bowel disease during pregnancy and postpartum. Am J Gastroenterol. 1981 Apr;75(4):265–9.
44. Khalili H, Higuchi LM, Ananthakrishnan AN, Richter JM, Feskanich D, Fuchs CS, et al. Oral contraceptives, reproductive factors and risk of inflammatory bowel disease. Gut. 2013 Aug;62(8):1153–9.
45. Ortizo R, Lee SY, Nguyen ET, Jamal MM, Bechtold MM, Nguyen DL. Exposure to oral contraceptives increases the risk for development of inflammatory bowel disease: a meta-analysis of case-controlled and cohort studies. Eur J Gastroenterol Hepatol. 2017 Sep;29(9):1064–70.
46. Looijer-van Langen M, Hotte N, Dieleman LA, Albert E, Mulder C, Madsen KL. Estrogen receptor-β signaling modulates epithelial barrier function. Am J Physiol Gastrointest Liver Physiol. 2011 Apr;300(4):G621–6.
47. Principi M, Barone M, Pricci M, De Tullio N, Losurdo G, Ierardi E, et al. Ulcerative colitis: from inflammation to cancer. Do estrogen receptors have a role? World J Gastroenterol. 2014 Sep 7;20(33):11496–504.
48. Wada-Hiraike O, Imamov O, Hiraike H, Hultenby K, Schwend T, Omoto Y, et al. Role of estrogen receptor beta in colonic epithelium. Proc Natl Acad Sci U S A. 2006 Feb 21;103(8):2959–64.
49. Ichimiya T, Yamakawa T, Hirano T, Yokoyama Y, Hayashi Y, Hirayama D, et al. Autophagy and Autophagy-Related Diseases: A Review. Int J Mol Sci. 2020 Nov 26;21(23):8974.
50. Shao BZ, Yao Y, Zhai JS, Zhu JH, Li JP, Wu K. The Role of Autophagy in Inflammatory Bowel Disease. Front Physiol. 2021 Feb 3;12:621132.
51. Jacenik D, Cygankiewicz AI, Mokrowiecka A, Małecka-Panas E, Fichna J, Krajewska WM. Sex- and Age-Related Estrogen Signaling Alteration in Inflammatory Bowel Diseases: Modulatory Role of Estrogen Receptors. Int J Mol Sci. 2019 Jun 28;20(13):3175.
52. Huang Z, Chen B, Liu X, Li H, Xie L, Gao Y, et al. Effects of sex and aging on the immune cell landscape as assessed by single-cell transcriptomic analysis. Proc Natl Acad Sci U S A. 2021 Aug 17;118(33):e2023216118.
53. Afshan G, Afzal N, Qureshi S. CD4+CD25(hi) regulatory T cells in healthy males and females mediate gender difference in the prevalence of autoimmune diseases. Clin Lab. 2012;58(5-6):567–71.