Abstract
When immune cells are activated, they undergo metabolic change in order to have sufficient energy to function effectively. The Krebs cycle is one of the most important pathways involved in this response and citrate, a critical component of this pathway, regulates carbohydrate and lipid metabolism. Recently, citrate has also emerged as a key immunometabolite, exerting specific effects on immunity and inflammation. Citrate has been shown to be involved in the production of prostaglandins and nitric oxide, and via the generation of malonyl-CoA and acetyl-CoA in the cytosol, to impact cytokine production. Citrate can also generate itaconate via aconitate, which promotes an anti-inflammatory response. All these findings support citrate as a crucial signal for immune cell activation and function.
In this review we will describe the various ways through which citrate participates in immune cell behaviour and speculate on the targeting of citrate as a possible novel therapeutic approach for immune and inflammatory diseases.
Keywords
Citrate; Krebs cycle; immunometabolism; itaconate; inflammation; ATP-citrate lyase
Introduction
Citrate, or citric acid, is one of the most common acids in plants and a metabolic product in aerobic organisms. Beyond its reputation for its anticoagulant activity, first described in the early 1990s [1], citrate is well known as a master metabolite in living cells.
Citrate is one of the most important intermediates in Krebs Cycle (also called the citric acid cycle) and has several roles depending on whether it remains inside mitochondria or is released into the cytosolic compartment [2].
It is a fundamental intermediate in aerobic nutrientderived energy production and it was subsequently shown to link carbohydrate metabolism to fatty acid metabolism [3].
Much more recently, citrate is generating much interest in the areas of immunometabolism and immune cell activation [4]. Here, we explore the role of citrate in inflammation and how it can modulate immune cell responses via multiple mechanisms. In many ways, citrate has been reborn as a metabolite critical for the immune system.
Krebs cycle
Krebs cycle, elucidated by Sir Hans Krebs, is a bioenergetic pathway occurring in the mitochondrial matrix of all eukaryotic organisms. It is an essential process for energy generation from diet carbon-based nutrients. Krebs was studying how bird muscle tissue takes up oxygen very quickly in the presence of carbohydrates and hypothesized that carbohydrate metabolism should occur through a cyclic series of reactions [5].
Glucose and lipid catabolism-derived acetyl-CoA is redirected to the Krebs cycle in order to generate ATP and redox coenzymes. These redox cofactors will be then used to produce the energy necessary for the cell through the mitochondrial electron transport chain, during a process known as oxidative phosphorylation (OXPHOS).
We now know however that Krebs cycle rewiring occurs in activated immune cells and is pivotal for protection against viral and bacterial infection. This process, known as metabolic reprogramming [6], is essential for the production of energy and metabolites that can also act as immune signalling molecules.
Early metabolomic studies on macrophages activated with the Gram negative bacterial product LPS revealed that Krebs cycle undergoes fragmentation at two important points; these breaks lead to the withdrawal of metabolic intermediates from the cycle [3,7,8]. A breakpoint at SDH (succinate dehydrogenase) leads to succinate accumulation which promotes HIF1α stabilization and boosts the expression of inflammatory genes such as that encoding IL1-β.
Succinate oxidation via SDH however can also boost HIF1-α via reactive oxygen species (ROS) generated by mitochondrial complex I, indicating complexity in the role of succinate regarding HIF1-α [3]. The second breakpoint occurs at the level of isocitrate dehydrogenase (IDH), which is responsible for the conversion of isocitrate into α-ketoglutarate [9]. This block will lead to citrate accumulation which has various downstream consequences for inflammatory cell activation.
Krebs cycle as the only source of endogenous citrate has been considered so far and if microorganism infections could somehow boost or impair citrate uptake from the extracellular environment is a question that still need to be answered.
Citrate as an immunomodulator: the role of CIC
Few studies have actually received attention about the role of citrate during viral and bacterial infections in animal cells. Citrate salts were previously used to protect plants against phytopathogens while reducing the use of pesticides. For example, citrate has been shown to protect lettuce from four different species of Shigella [10]. Important evidence of a role for citrate in blocking microbial infection in mammals has also been indicated in a study by Hojo et al., who found that oral bacteria such as Fusobacterium Nucleatum, Streptococcus Mutans and Streptococcous Pneumoniae are susceptible to sodium citrate, although the underlying mechanisms have not been defined yet [11]. Citrate also inhibits the growth of the Gram negative bacteria Arcobacter butzleri [12] thanks to its acid pH and has antibacterial activity also against Gram positive Staphylococci, including MRSA strains [13].
As regarding citrate role during viral infection, the iron salt ferric ammonium citrate (FAC) blocked Influenza A virus, ZIKA virus, HIV virus and Enterovirus 71 (EV71) infections [14]. In vitro, in A549 human lung epithelial cell line and in human monocytic THP-1 macrophages, FAC inhibits IAV PR8 infection and virus-induced IL1-β production [14]. In vivo, mice treated with FAC after IAV PR8 airways injection significantly blocked viral replication dampening IL1-β, IFN-β, CCL2, IL-6 and TNF-α expression, decreasing the number of migrated neutrophils to the lungs and lengthening mice survival [14]. FAC treatment had a significant effect also on HIV- 1 through the inhibition of infection in human PBMCdifferentiated dendritic cells and the related virus-induced immune response mediated by IL-6, IFN-β and IL1-β [14]. Moreover, Vero cells infected with Zika virus showed low viral RNA 72 hours post FAC treatment while mice injected with the same virus displayed decreased weight loss and longer survival upon ferric citrate supplement [14]. Interestingly, FAC decreased EV71 replication in THP-1 macrophages, thus leading to inhibition of NLRP3 inflammasome-dependent Caspase-1 and IL1-β release [14]. Indeed, FAC impaired the induction of the oligomerization of NLRP3 inflammasome adapter protein ASC [14].
Regarding the immunomodulatory role of citrate, most of the work has been carried out on macrophages specifically exploring the effect of LPS. In THP-1 cells at high concentrations, when calcium availability was limiting due to sequestration by citrate chelation, citrate inhibited TNF-α and IL-8 mRNA production following LPS stimulation [15]. However, when calcium availability was not limiting, as can be seen with low citrate concentrations, citrate augmented TNF-α and IL-8 mRNA production [15]. Indeed, citrate treatment promoted histone H3 and H4 acetylation because of acetyl-CoA production by ATP citrate lyase (ACLY) (Figure 1). H3 lysine 9 acetylation, H4 lysine 8 acetylation and H4 lysine 12 acetylation were augmented significantly in THP-1 cells incubated with citrate compared to controls at both the TNF-α and IL-8 promoter regions [15]. This demonstrates how citrate drives inflammatory gene expression via histone acetylation. Ji-Sook et al. described the use of calcium citrate, the calcium salt of citric acid, on the mouse macrophage cell line RAW 264.7 cells [16]. Calcium citrate strongly reduced LPS-induced ROS generation and rescued the activity of antioxidant defence enzymes such as Superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) (Figure 2) [16]. Exposure to LPS enhanced NO production and IL-6, IL-1β and TNF-α expression, which were all suppressed by calcium citrate treatment. NF-κB, iNOS and COX-2 expression were augmented after LPS immune cells activation and then significantly inhibited by calcium citrate (Figure 2) [16]. These early studies highlighted the difficulties of understanding the role of citrate in macrophages.
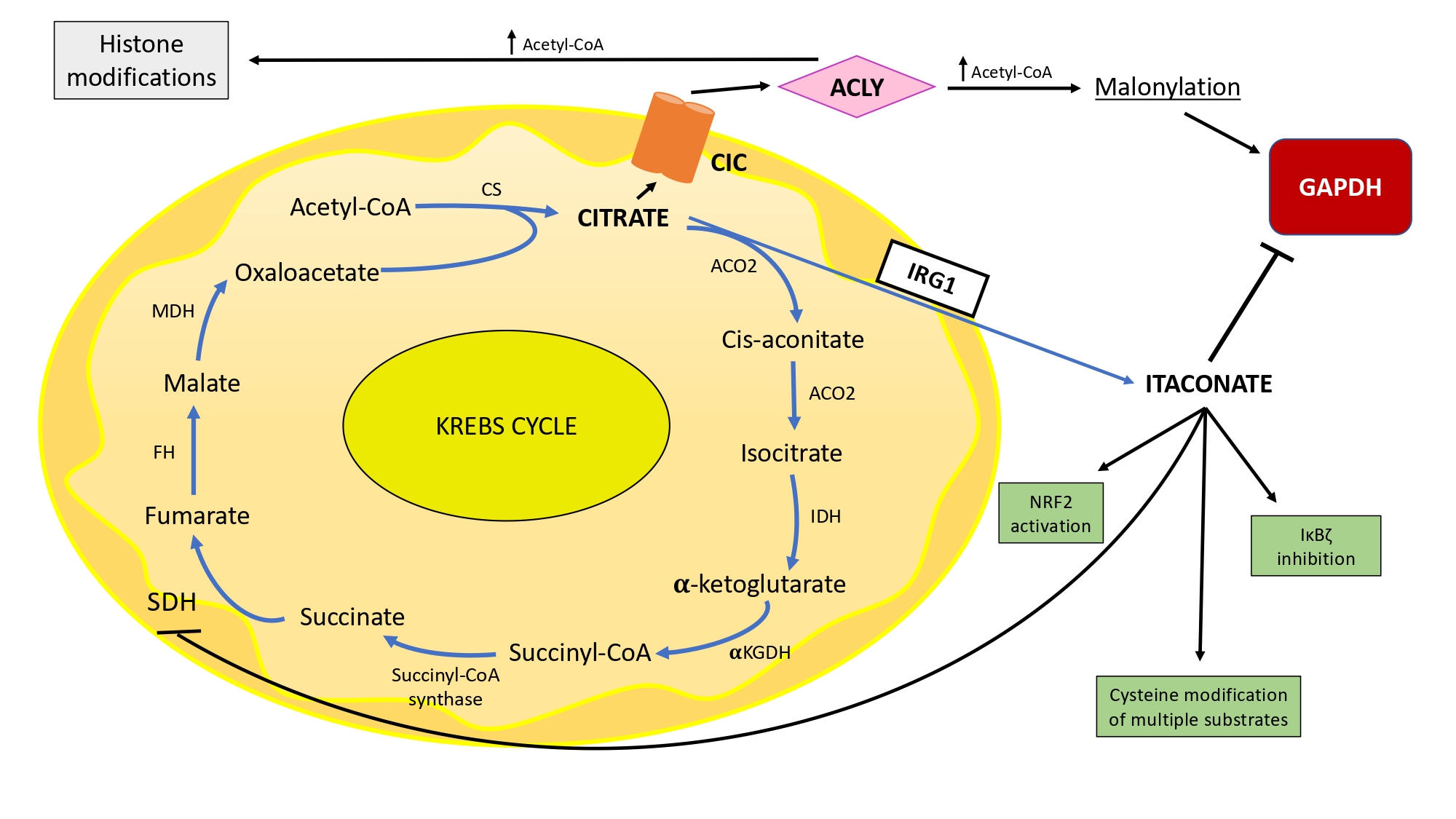
Citrate generates itaconate through IRG1 decarboxylase. Outside mitochondria, itaconate inhibits GAPDH and SDH activity and drives Nrf2 activation and IkBζ inhibition. Itaconate also modifies multiple target proteins on Cysteine (ex. LDH-A and GAPDH) which might also contribute to its anti-inflammatory effect.
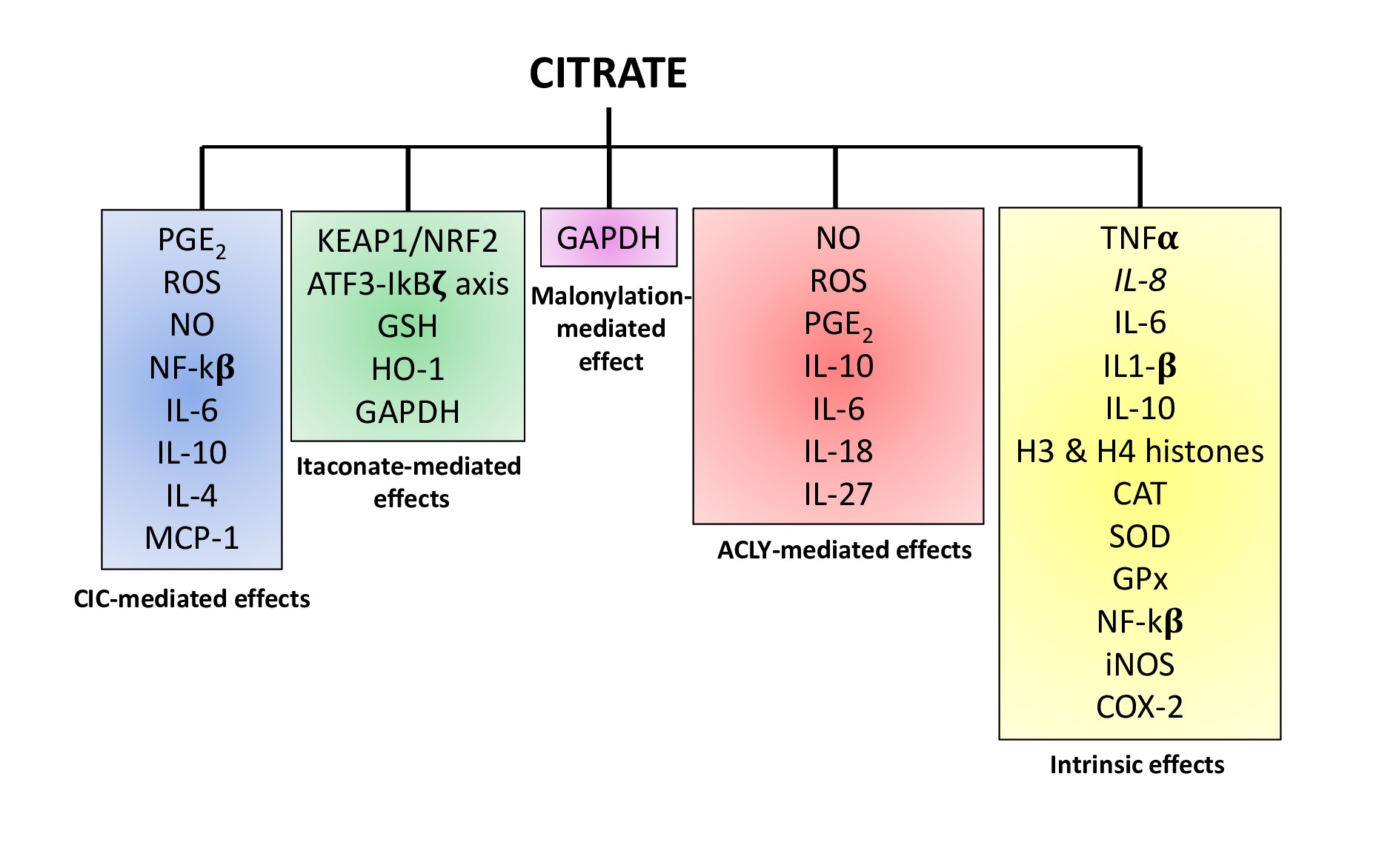
Citrate plays an immunomodulatory role through the IRG1-dependent production of itaconate and its transmembrane carrier CIC. Its availability in the cytosol provides acetyl-CoA pool for ACLY-mediated and malonylation-mediated regulation of genes and proteins involved in inflammation signaling and oxidative stress development, such as IL1-β, COX-2, NO and PGE2.
The first study clearly pointing to a role for citrate in macrophage activation was published by Infantino et al. [17] in 2011 and showed for the first time citrate carrier (CIC or also called SLC25A1) involvement in inflammation. CIC transports citrate from the mitochondria in exchange for cytosolic malate. Infantino demonstrated that CIC makes citrate available for fatty acid-dependent generation of lipid inflammatory modulators like prostaglandins (PGs) [17]. CIC mRNA levels were increased in LPS-stimulated macrophages while CIC silencing or inhibition in activated macrophages led to a significant decrease in the amount of NO, PGE2 and ROS. An in silico analysis for the human CIC gene promoter found two NF-κB-responsive elements [17]. All these outcomes propose CIC as a NF-κB target gene required for LPS-driven inflammation through citrate pool availability.
The key role of CIC was confirmed in human macrophage cell line U937 treated with TNFα and IFN-γ alone or in combination showing an upregulation of CIC mRNA [18]. Inhibition of IκB kinase, whose activity is required for NF- κB function, or the use of p50-specific antibody reduced CIC mRNA and protein levels after TNFα stimulation [18]. This indicated a NF-κB dependent mechanism for TNFα-driven citrate carrier induction. Concurrently, the use of a STAT inhibitor strongly decreased CIC mRNA and protein and IFN-γ, alone or in combination, there was a prominent compared to cells only treated with IFN-γ and showed that CIC is boosted by IFN-γ through a STAT-dependent mechanism [18]. In CIC-silenced cells induced with TNF-α decrease in PGE2, NO and ROS levels (Figure 2) [18]. This work displays how CIC is responsible for pro-inflammatory mediator production after cytokine-mediated activation of macrophages.
A recent publication by Avantaggiati et al. [19] might shed light on a possible mechanism for citrate contribution to inflammatory disease pathogenesis. The role of mitochondrial citrate carrier CIC was examined in nonalcoholic fatty liver disease (NAFLD) and the development of inflammatory steatohepatitis (NASH). Impairment of CIC by an inhibitor called CTPI-2, which competitively binds to its citrate-binding pocket, or complete knockout (KO) of the CIC gene lead to lowering of IL-6 and monocytes chemoattractant protein-1 (MCP1) levels and increased IL-10 and IL-4 [19]. Liver tissue resident macrophages, also known as Kupffer cells, were highly increased following a high fat diet (HFD) in mice and the positive trend was reversed by the use of CTPI-2. CIC inhibition significantly blocked inflammatory M1 macrophage polarization and tissue infiltration, as evidenced by TNF-α and iNOS expression [19]. Alternative pathways that could compensate for citrate deficiency involve isocitrate dehydrogenases 1 and 2 (IDH 1 and 2) and acetyl-CoA synthetases (ACSS 1 and 2), that synthetize acetyl-CoA from acetate. CTPI-2 reduced both of these enzymes and increased mRNA and protein expression of SLC13A5, a transporter which takes up citrate from the extracellular environment [19]. This showed that extracellular citrate can be the only source of cytosolic acetyl-coA in absence of CIC.
CIC is thus essential for the role of citrate as a signal in immune cells and, hence, it could be a candidate target for treatment of immune diseases. Counteracting CIC activity through CIC inhibitors or CIC silencing could induce an anti-inflammatory and resolution response while dampening local inflammation development.
Citrate and ACLY
Beyond citrate transport, another process that has been shown to be important for citrate in inflammation involves the enzyme ATP-dependent citrate lyase (ACLY). ACLY promotes citrate conversion to acetyl-CoA and oxaloacetate outside mitochondria. It drives acetyl-CoA pool availability and sustains cell activation in innate and adaptive immunity (Figure 1) [3]. The Infantino study highlighted the importance of citrate export from the mitochondria for its immunomodulatory effects. This has been confirmed in studies on ACLY which has been implicated in LPS-induced epigenetic changes [20]. The resulting acetyl-CoA can feed the mevalonate pathway for cholesterol formation, promote fatty acid synthesis for membrane biogenesis or be a substrate for acetylation of proteins including histones, thus participating in epigenetic modifications [3,20]. ACLY guides acetylation in Toll like receptor (TLR)-dependent gene expression via acetyl-CoA derived from citrate [19].
It is well known that genes encoding for inflammatory mediators are epigenetically regulated via acetylation and methylation in their promoter regions and on histones [21-23], thus driving silencing or activation of transcription. The Infantino et al. study had underlined the indispensable role of ACLY in macrophages activation, during which LPS, IFNγ and TNFα up-regulate ACLY protein and mRNA expression [24]. It has also been shown that the inhibition of ACLY by radicicol decreased NO, ROS and PGE2 production in activated THP- 1 cells (Figure 2) [24], pointing to its role in modulating inflammatory mediator production.
ACLY induction during macrophage activation was confirmed in 2019 by Lauterbach et al., who examined the LPS-TLR4 axis in macrophage metabolism and histones acetylation. They found that LPS increased the extracellular acidification rate (ECAR) in bone marrowderived macrophages within 2 hours after stimulation, thus increasing glycolysis [25]. LPS signalling resulted in increased maximal respiration in macrophages when measured 8 hours after LPS exposure. In contrast, LPS activation of BMDMs for 24 hours led to decreased basal OXPHOS as measured by oxygen consumption rate (OCR) [25]. These data suggested that mitochondrial metabolism reacts rapidly to TLR4 signalling and that the early response, relevant for gene transcription, differed from that obtained after 24 hours of LPS stimulation [25].
LPS increased CXCL1, IL-6, IL-1β expression and synthesis of some important metabolites of the Krebs cycle such as succinate, lactate and citrate. The change in citrate encouraged them to analyse ACLY expression, which was found to be induced through a MyD88 and TRIFdependent mechanism [25]. RNA-seq of LPS-stimulated BMDMs treated with ACLY inhibitor showed increased mRNA expression of IL-10 and IL-1R antagonist (IL1- RA) and decreased mRNA expression of IL-6, IL-18, IL- 27, identified as ACLY target genes regulated by histone modification (Figure 2) [25].
ACLY can be in turn regulated by acetylation [26] and phosphorylation by Akt [27]. The latter is part of IL-4 mediated M2 macrophages activation. IL-4 treatment of BMDMs increased glucose uptake and glycolysis and induced expression of M2 genes Arg1, Retnla, Mgl2 [27]. The use of an Akt inhibitor reduced Arg1, Retnla and Mgl2 induction, suggesting an Akt-dependent mechanism for M2 polarization [27]. To understand how Akt regulates M2 genes, histones acetylation in IL-4 treated macrophages demonstrated that IL4-R axis enhanced acetylation of H3 and H4 histones while the Akt inhibitor strongly decreased it. IL-4 stimulation promoted the phosphorylation of ACLY by Akt, thus leading to M2 gene expression [27]. Indeed, ACLY inhibition abolished Akt-dependent M2 polarization, identifying ACLY as a master regulator for M2 activation.
Another study by Dekoter et al. revealed a new role for ACLY during macrophage differentiation. Myeloid progenitor cells differentiate into macrophages and upregulate the transcriptional factor PU.1, essential for their development [28]. The induction of PU.1 expression in BN cells, myeloid precursor cells lacking PU.1 factor, downregulated mRNA levels of ACLY and reduced cell cycle progression. The same downregulation was observed also in M-CSF-dependent differentiation of bone marrow cells [28]. ACLY inhibitor treatment on cultured BN cells abolished cell proliferation, indicating ACLY as the enzyme involved in cell cycle promotion. These outcomes led to study on whether acetate or acetyl-CoA supplementation could rescue cell cycle progression. Both metabolites increased cell cycle progression of BN cells and suggested that ACLY-dependent synthesis of acetyl-CoA is required for macrophage maturation [28].
Apart from macrophages, ACLY expression has been detected in T and B lymphocytes. Bacterial LPS induces ACLY expression and enzymatic activity in naïve B lymphocytes, thus driving their differentiation into Igsecreting plasma cells through the support of glucosedependent de novo lipogenesis aimed at expanding the lymphocyte endomembrane network [29]. Moreover, ACLY phosphorylation on Ser455 by Akt in T lymphocytes is induced by IL-2 and promotes acetylation of cell cycle regulating genes thereby driving to CD4+ T cells proliferation [30].
All these studies point to ACLY as a key driver for innate and adaptive immune cell activation via its role in promoting acetylation. Given the role of ACLY in immune cells, its inhibition might have potential as an antiinflammatory therapeutic. In vivo studies are necessary to thoroughly characterize this enzyme and assess if its expression could be modulated by citrate itself. We know from previous literature that homozygous Acly knockout mice died early in development. So heterozygous mice for ACLY can be a suitable model for studying its therapeutic potential.
Post-translational modifications caused by citrate
Post-translational modifications (PTMs) are reversible modifications of specific aminoacids that can change the tertiary structure and consequent function of proteins. Acetyl-CoA provokes acetylation, a PTM that prominently occurs on lysine on histones [4]. Histones can also receive an acetyl group from substrates such as malonyl-CoA, succinil-CoA, crotonil-CoA, glutaryl-CoA, to as lysine acylation [31].
Apart from acetylation, malonylation is another posttranslational modification which might regulate protein function [32]. This also depends on citrate, since malonyl- CoA is generated from acetyl-CoA, which as we saw in the preceding section comes from citrate (Figure 1). Malonyl- CoA is generated by acetyl-CoA carboxylase ACC1 (of which there are cytosolic and mitochondrial isoforms), while the reverse reaction is catalysed by malonyl-CoA decarboxylase (MCD) [33]. The major role of malonyl- CoA is as an intermediate in de novo fatty acid synthesis and elongation [34]. It blocks β-oxidation of fatty acids through allosteric inhibition of carnitine-palmitoyl transferase (CPT1) [35-37]. Lysine residues can however be modified by malonyl-CoA [32]. This makes Lysine residues negatively charged and SIRT5 can control this process by acting as a demalonylase [38-40].
The first report of malonylation as an inflammatory signal was shown in LPS treated BMDMs where it increased expression of ACC1 and mitochondrial malonyl- CoA synthetase 3 (ACSF3) [41]. Boosting malonyl-CoA production led to malonylation of many proteins and in particular of the glycolytic enzyme GAPDH protein on Lys 213 [41]. GAPDH is required for the induction of cytokines such as IL1-β and IL-6, but a more relevant role shows up in TNF-α modulation [41]. GAPDH has RNA-binding activity and binds to AU-rich elements in TNF-α mRNA in resting macrophages, thus blocking its translation [41]. LPS stimulation induces GAPDH malonylation leading to its dissociation from TNF-α mRNA, so that TNFα can be translated [41]. This suggests that inhibiting ACC1 (which also has a role in Th1 cell development) or ACSF3 (which also is toxic to mitochondria) could have anti-inflammatory potential.
It remains unknown whether malonylation affects other proteins involved in inflammatory signalling, apart from GAPDH. It is also unknown whether malonylation occurs when other inflammatory stimuli, such as TNF-α and IFN-γ, activate immune cells. ACC1 is one of the enzymes making malonyl-CoA available for fatty acid biosynthesis, an event known to be pro- inflammatory. Further studies should address whether ACC1 inhibition might modulate malonylation and consequently inflammatory response.
Upregulation of SIRT1 function might decrease citratemediated malonylation of proteins but specific enzymeamino acid interactions have to be sharply identified.
Citrate as a regulator of itaconate
A final recent aspect concerning citrate is its role in generating itaconate, which is derived from decarboxylation of cis-aconitate, the intermediate product of citrate conversion to isocitrate by aconitase (ACO2) during the second step of Krebs cycle [42]. The shift in Krebs cycle towards itaconate production is a prominent consequence of LPS-dependent activation in macrophages [43]. The enzyme responsible for itaconate generation, cisaconitate decarboxylase, is encoded by the IRG1 gene, first discovered in 1995 [44].
The first studies about itaconate action focus on its antimicrobial and growth inhibition activity towards various microorganisms [45-47]. A key target of itaconate was isocitrate lyase (ICL), the enzyme responsible for the cleavage of isocitrate into succinate and glyoxylate during the first step of the glyoxylate shunt, a process not present in mammals [46]. Itaconate was then shown to have a role in mammalian cells and in particular to exert an antiinflammatory effect. Itaconate production is induced in macrophages by LPS and IFNγ [48] and one of its targets is succinate dehydrogenase (SDH). This inhibits ROS generation from complex I, lowering IL1-β production in BMDMs [48].
Succinate levels in macrophages are strictly linked to itaconic acid metabolism. Itaconate reduces ROS/RNS (reactive nitrogen species) and improves neurological function in a model of ischemia-reperfusion confirming its role as a ROS regulator [49].
Another mechanism of action for itaconate is modulation of the transcriptional factor Nuclear factor erythroid 2-related factor 2 (NRF2) [50] (Figure 1), essential in driving anti-oxidant and anti-inflammatory responses [51], which in physiological conditions is controlled by Kelch-like ECH-associated protein 1 (KEAP1) [52]. A new post-translational modification – called 2,3-dicarboxypropylation - occurs on cysteines of KEAP1 in human and murine macrophages treated with the itaconate derivative 4-Octyl Itaconate (4-OI) [50]. This specific change inactivates KEAP1 making NRF2 free to translocate to the nucleus and induce transcription of antioxidant enzymes such as heme-oxygenase (HO-1) and glutathione (GSH) and repression of pro-inflammatory cytokines [50]. Endogenous itaconate is likely to regulate Nrf2 since this response was reduced in IRG1- deficient macrophages [52,53]. Multiple targets for 2,3- dicarboxypropylation were identified, notably of enzymes in glycolysis [50,54] but also inflammatory proteins such as NLRP3 and Gasdermins [55].
4-OI also modifies and inhibits GAPDH in LPS-stimulated RAW264.7 macrophages [56] (Figure 1), further indicating that GAPDH is a target for citrate-derived modification, as shown for malonylation (Figure 2). 4-OI alkylation of GAPDH on Cys22 via Michael addition inhibited its activity, hindering IL-1β and iNOS expression [56].
Furthermore, dimethyl itaconate (DMI) abolishes LPSdependent induction of IκBζ through ATF3 transcriptional factor (Figure 1) and blocks this axis responsible for secondary transcriptional responses in activated BMDMs [57]. DMI-treated bone marrow-derived macrophages (BMDMs) displayed upregulation of Nrf2 and its target genes Hmox1, Nqo1 and Gclm [57]. DMI structure was found to act as an electrophile in Michael reactions and trigger electrophilic stress. Usually, electrophilic stress affects the glutathione (GSH) pool. DMI treatment decreased GSH intracellular concentration and increased ROS cell production [57]. The transcription factor IκBζ regulates secondary transcriptional response to TLR activation. DMI treatment totally abolished the LPS-associated induction of IκBζ protein and inhibited its target genes, but did not change IκBζ mRNA expression, meaning that it has to be affected at the post-transcriptional level [57]. Indeed, electrophilic stress induced by DMI presence increased phosphorylation of macrophage eIF2α and consequently the inhibition of IκBζ translation [57]. RNAseq of DMI-treated WT and Nrf2−/− BMDMs showed an upregulation of ATF3 transcriptional factor. Atf3−/− cells restored IκBζ protein levels upon DMI treatment and significantly increased IL-6 production [57]. Complete deficiency for Atf3 in macrophages failed to increase eIF2α phosphorylation after DMI treatment, thus confirming the ATF3-dependent effect of DMI on IκBζ regulation [57].
These studies reveal citrate as a precursor for itaconate production and its several actions as a crucial immunomodulatory metabolite. Itaconate could be a potential therapeutic treatment for inflammatory disease in the future. Other studies are needed to assess the existence of a cell surface receptor for itaconate and to know if some of the effects previously reported could be receptor-mediated effects. Moreover, itaconate could even boost a positive or negative feedback loop on citrate production and transport. Overexpression of Irg1, the enzyme generating itaconate, could lead to a rise in itaconate production and a subsequent activation of cellular anti-oxidant and antiinflammatory response.
Concluding Remarks
In this review, we have discussed the role of citrate as a critical signal in immunity and inflammation. LPSinduced metabolic reprogramming in immune cells drives citrate pool availability and CIC and ACLY expression. A breakpoint across Krebs cycle leads to citrate withdrawal from the cycle and production of itaconate, an antiinflammatory metabolite.
Outside mitochondria, ACLY generates acetyl-CoA which leads to histone modification and malonylation of proteins. Thus, citrate represents a key metabolite in various effector response in immune cells, although its mechanisms of action need to be further defined.
Here, we consider citrate as a Krebs cycle intermediate and we do not know how inflammatory triggers would modulate its uptake from the extracellular environment. Citratedependent modulation of its carrier for extracellular uptake is a subject that would be fascinating to explore.
Citrate pathway investigation in tissue resident immune cells such as tissue resident macrophages and DCs could unveil how tissue specific microenvironment drive citrate production and would be helpful to gain further insight into leukocytes characterization and study of local inflammatory response.
Despite its clear immunomodulatory effects, further studies have to be carried out in order to understand which will be citrate impact on other fundamental immunometabolites production in mitochondria and if it could in turn impair Krebs cycle.
The use of a citrate synthase knockout would stop citrate production and prevent all these citratemediated mechanisms, potentially confirming its role in inflammation. Although this would also impair Krebs cycle. Additional in vivo models would be useful to discover how the citrate pathway can be used as a therapeutic target for inflammatory diseases. Studies into some variations of the CIC gene and susceptibility to inflammatory disease might prove informative.
We can look forward to further insights into the role of citrate and its metabolism in immunity and inflammation in both health and disease.
Authors Contribution
AZ and LAO’N wrote the manuscript. ZZ corrected the draft and gave advice on the manuscript. LAO’N supervised and edited the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
2. Wilson BA, Schisler JC, Willis MS. Sir Hans Adolf Krebs: Architect of Metabolic Cycles. Laboratory Medicine. 2010 Jun 1;41(6):377-80.
3. Williams NC, O’Neill LAJ. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Frontiers in Immunology. 2018 Feb 5;9(141).
4. O’Neill LA. A critical role for citrate metabolism in LPS signalling. Biochemical Journal. 2011 Sep 15;438(3):e5-6.
5. Kornberg H. Krebs and his trinity of cycles. Nature Reviews Molecular Cell Biology. 2000 Dec;1(3):225-8.
6. Ryan DG, O’Neill LAJ. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Letters. 2017 Oct 1;591(19):2992-3006.
7. Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015 Mar 17;42(3):419-30.
8. Tannahill GM, Curtis AM, Adamik J, Palsson- McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1β through HIF1α. Nature. 2013 Apr;496(7444):238-42.
9. Akram M. Citric acid cycle and role of its intermediates in metabolism. Cell Biochemistry and Biophysics. 2014 Apr 1;68(3):475-8.
10. In Y-W, Kim J-J, Kim H-J, Oh S-W. Antimicrobial Activities of Acetic Acid, Citric Acid and Lactic Acid against Shigella Species. Journal of Food Safety. 2013 Feb;33(1):79-85.
11. Nagaoka S, Murata S, Kimura K, Mori T, Hojo K. Antimicrobial activity of sodium citrate against Streptococcus pneumoniae and several oral bacteria. Letters in Applied Microbiology. 2010 Nov;51(5):546-51.
12. Phillips CA. The effect of citric acid, lactic acid, sodium citrate and sodium lactate, alone and in combination with nisin, on the growth of Arcobacter butzleri. Letters in Applied Microbiology. 1999 Dec;29(6):424-8.
13. Lee Y-L, Thrupp L, Owens J, Cesario T, Shanbrom E. Bactericidal activity of citrate against Gram-positive cocci. Letters in Applied Microbiology. 2001 Nov 6;33(5):349- 51.
14. Wang H, Li Z, Niu J, Xu Y, Ma L, Lu A, et al. Antiviral effects of ferric ammonium citrate. Cell Discovery. 2018 Mar 27;4(1):1-14.
15. Ashbrook MJ, McDonough KL, Pituch JJ, Christopherson PL, Cornell TT, Selewski DT, et al. Citrate modulates lipopolysaccharide-induced monocyte inflammatory responses. Clinical & Experimental Immunology. 2015 Jun;180(3):520-30.
16. Choi EY, Kim HJ, Han JS. Anti-inflammatory effects of calcium citrate in RAW 264.7cells via suppression of NF-κB activation. Environmental Toxicology and Pharmacology. 2015 Jan 1;39(1):27-34.
17. Infantino V, Convertini P, Cucci L, Panaro MA, Di Noia MA, Calvello R, et al. The mitochondrial citrate carrier: a new player in inflammation. Biochemical Journal. 2011 Sep 15;438(3):433-6.
18. Infantino V, Iacobazzi V, Menga A, Avantaggiati ML, Palmieri F. A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochimica et Biophysica Acta (BBA)-Gene Regulatory Mechanisms. 2014 Nov 1;1839(11):1217-25.
19. Tan M, Mosaoa R, Graham GT, Kasprzyk-Pawelec A, Gadre S, Parasido E, et al. Inhibition of the mitochondrial citrate carrier, Slc25a1, reverts steatosis, glucose intolerance, and inflammation in preclinical models of NAFLD/NASH. Cell Death & Differentiation. 2020 Jan 20:1-5.
20. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP- citrate lyase links cellular metabolism to histone acetylation. Science. 2009 May 22;324(5930):1076-80.
21. Daskalaki MG, Tsatsanis C, Kampranis SC. Histone methylation and acetylation in macrophages as a mechanism for regulation of inflammatory responses. Journal of Cellular Physiology. 2018 Sep;233(9):6495- 507.
22. Li T, Garcia-Gomez A, Morante-Palacios O, Ciudad L, Özkaramehmet S, Van Dijck E, et al. SIRT1/2 orchestrate acquisition of DNA methylation and loss of histone H3 activating marks to prevent premature activation of inflammatory genes in macrophages. Nucleic Acids Research. 2020 Jan 24;48(2):665-81.
23. Klein K, Gay S. Epigenetics in rheumatoid arthritis. Current Opinion in Rheumatology. 2015 Jan 1;27(1):76- 82.
24. Infantino V, Iacobazzi V, Palmieri F, Menga A. ATPcitrate lyase is essential for macrophage inflammatory response. Biochemical and Biophysical Research Communications. 2013 Oct 11;440(1):105-11.
25. Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll- like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity. 2019 Dec 17;51(6):997-1011.
26. Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, et al. Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Molecular Cell. 2013 Aug 22;51(4):506-18.
27. Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, et al. Akt- mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife. 2016 Feb 19;5:e11612.
28. Rhee J, Solomon LA, DeKoter RP. A role for ATP Citrate Lyase in cell cycle regulation during myeloid differentiation. Blood Cells, Molecules, and Diseases. 2019 May 1;76:82-90.
29. Dufort FJ, Gumina MR, Ta NL, Tao Y, Heyse SA, Scott DA, et al. Glucose-dependent de novo lipogenesis in B lymphocytes: a requirement for atp-citrate lyase in lipopolysaccharide-induced differentiation. Journal of Biological Chemistry. 2014 Mar 7;289(10):7011-24.
30. Osinalde N, Mitxelena J, Sánchez-Quiles V, Akimov V, Aloria K, Arizmendi JM, et al. Nuclear Phosphoproteomic Screen Uncovers ACLY as Mediator of IL-2-induced Proliferation of CD4+ T lymphocytes. Molecular & Cellular Proteomics. 2016 Jun 1;15(6):2076-92.
31. Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nature reviews Molecular cell biology. 2017 Feb;18(2):90- 101.
32. Hirschey MD, Zhao Y. Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Molecular & Cellular Proteomics. 2015 Sep 1;14(9):2308-15.
33. Trefely S, Lovell CD, Snyder NW, Wellen KE. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Molecular Metabolism. 2020 Feb 14:100941.
34. Saggerson D. Malonyl-CoA, a key signaling molecule in mammalian cells. Annual Review of Nutrition. 2008 Aug 21;28:253-72.
35. Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. The Journal of Clinical Investigation. 2012 Jun 1;122(6):1958-9.
36. Karwi QG, Jörg AR, Lopaschuk GD. Allosteric, transcriptional and post-translational control of mitochondrial energy metabolism. Biochemical Journal. 2019 Jun 28;476(12):1695-712.
37. Miotto PM, Steinberg GR, Holloway GP. Controlling skeletal muscle CPT-I malonyl- CoA sensitivity: the importance of AMPK-independent regulation of intermediate filaments during exercise. Biochemical Journal. 2017 Feb 15;474(4):557-69.
38. Gorska M, Dobrzyn A, Baranowski M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Medical Science Monitor. 2005 Jan 1;11(1):CR35-8.
39. Nishida Y, Rardin MJ, Carrico C, He W, Sahu AK, Gut P, et al. SIRT5 Regulates both Cytosolic and Mitochondrial Protein Malonylation with Glycolysis as a Major Target. Molecular Cell. 2015 Jul 16;59(2):321-32.
40. Peng C, Lu Z, Xie Z, Cheng Z, Chen Y, Tan M, et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Molecular & Cellular Proteomics. 2011 Dec 1;10(12):M111.012658.
41. Galván-Peña S, Carroll RG, Newman C, Hinchy EC, Palsson-McDermott E, Robinson EK, et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nature Communications. 2019 Jan 18;10(1):338.
42. Yu XH, Zhang DW, Zheng XL, Tang CK. Itaconate: an emerging determinant of inflammation in activated macrophages. Immunology and Cell Biology. 2019 Feb;97(2):134-41.
43. O’Neill LAJ, Artyomov MN. Itaconate: the poster child of metabolic reprogramming in macrophage function. Nature Reviews Immunology. 2019 May;19(5):273-81.
44. Lee CG, Jenkins NA, Gilbert DJ, Copeland NG, O’Brien WE. Cloning and analysis of gene regulation of a novel LPS-inducible cDNA. Immunogenetics. 1995 Mar 1;41(5):263-70.
45. Bellion E, Kelley RL. Inhibition by itaconate of growth of methylotrophic bacteria. Journal of bacteriology. 1979 May 1;138(2):519-22.
46. McFadden BA, Purohit S. Itaconate, an isocitrate lyase-directed inhibitor in Pseudomonas indigofera. Journal of Bacteriology. 1977 Jul 1;131(1):136-44.
47. Williams JO, Roche TE, McFadden BA. Mechanism of action of isocitrate lyase from Pseudomonas indigofera. Biochemistry. 1971 Apr 1;10(8):1384-90.
48. Strelko CL, Lu W, Dufort FJ, Seyfried TN, Chiles TC, Rabinowitz JD, et al. Itaconic acid is a mammalian metabolite induced during macrophage activation. Journal of the American Chemical Society. 2011 Oct 19;133(41):16386-9.
49. Cordes T, Lucas A, Divakaruni AS, Murphy AN, Cabrales P, Metallo CM. Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Molecular Metabolism. 2020 Feb 1;32:122-35.
50. Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018 Apr;556(7699):113-7.
51. Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2017 Feb 1;1863(2):585-97.
52. Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, et al. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino- terminal Neh2 domain. Genes & Development. 1999 Jan 1;13(1):76-86.
53. Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell metabolism. 2016 Jul 12;24(1):158-66.
54. Qin W, Qin K, Zhang Y, Jia W, Chen Y, Cheng B, et al. S-glycosylation-based cysteine profiling reveals regulation of glycolysis by itaconate. Nature Chemical Biology. 2019 Oct;15(10):983-91.
55. Qin W, Zhang Y, Tang H, Liu D, Chen Y, Liu Y, et al. Chemoproteomic Profiling of Itaconation by Bioorthogonal Probes in Inflammatory Macrophages. Journal of the American Chemical Society. 2020 Jun 4;142(25):10894-8.
56. Liao ST, Han C, Xu DQ, Fu XW, Wang JS, Kong LY. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nature communications. 2019 Nov 8;10(1): 5091.
57. Bambouskova M, Gorvel L, Lampropoulou V, Sergushichev A, Loginicheva E, Johnson K, et al. Electrophilic properties of itaconate and derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature. 2018 Apr;556(7702):501-4.