Abstract
The cytokine interlukin-6 (IL-6) is a vital mediator of the innate immune response, and a pleiotropic regulator of cellular function. Its involvement in autoimmune and inflammatory diseases, and cancer is illustrated by the clinical benefits of IL-6 blockade. Thus, there is a necessity to control the IL-6 production at transcriptional and post-transcriptional level. Post-transcriptional regulation that controls mRNA stability and translation appears to play a critical role in the regulation of not only Il6 expression but also Stat3, Ox40, T-bet and IL-17-induced expression of mRNAs by RNA-binding proteins. Few years ago, our group has reported a novel role of AT-rich interactive domain-containing protein 5A (Arid5a) in the post-transcriptional regulation of Il6 mRNA via binding to its 3’UTR thereby augmenting autoimmune disease, EAE. However, Regnase-1, an RNase, degrades mRNAs of multiple inflammatory genes such as Il6, Stat3, Ox40 etc. and contributes in the regulation of their mRNA stability by binding to 3’UTR of mRNAs. We found that Arid5a binds to the same region on 3’UTRs where Regnase-1 binds, and interferes Regnase-1 function thus, exacerbates autoimmune and inflammatory diseases. Briefly, our laboratory has identified several inflammatory mRNAs as targets of Arid5a and its contribution in many autoimmune and inflammatory disease such as experimental autoimmune encephalitis, sepsis, rheumatoid arthritis, acute lung injury etc. Furthermore, recent studies have shown Arid5a contribution in tumorigenesis too such as in lung cancer. Here, we highlight some recent progresses in Arid5a research to better understand Arid5a-mediated molecular mechanisms, and their relevance to Arid5a as promising therapeutic agent in immunological disorders.
Keywords
Posttranscriptional regulation, Arid5a, IL-6, Inflammation, Immunity
Commentary
The immune system is essential to host defense because it senses attacking pathogens and elicits protective immune responses. Although immune responses can protect against pathogens, uncontrolled immune responses cause tissue damage and other pathological consequences through their inflammatory mediators. For example, unsolicited or exaggerated IL-6 production causes severe inflammatory autoimmune diseases and cancer [1-3] and, thereby, Il6 expression is tightly regulated, both transcriptionally and post transcriptionally. RNA-binding proteins (RBPs) are known to regulate gene expression at the posttranscriptional level and are highly recognized owing to their associations with many biological functions and diseases [4-7]. AT-rich interactive domain-containing protein 5A (Arid5a) is known to be a cofactor of various transcription factors such as Sox9, and it also contributes to cell growth and differentiation [1,3,8]. Interestingly, our group has identified a novel function of Arid5a in mRNA stabilization as well as its associations with inflammation and autoimmunity [9-11]. We observed that chlorpromazine (CPZ), which is an antihistaminic and antipsychotic drug, inhibits lipopolysaccharide (LPS)-induced IL-6 production in macrophages [9,12]. Mechanistically, Arid5a, which is also inhibited by CPZ, functions in mRNA stability by binding to the adenylate/ uridylate (AU)- and U-rich elements (AREs) on the 3′ untranslated region (3′UTR) of the Il6 mRNA transcript, thereby facilitating the posttranscriptional regulation of Il6 gene expression, which in turn, increases the overproduction of IL-6 in vivo. Arid5a resides in the nucleus under normal conditions where it binds to various TFs, such as Sox9, to contribute to nuclear functions [8]. Subsequently, we have recently revealed that the Arid5a protein regulates the adipogenesis and the development of obesity in mice [13]. This study explained that Arid5a inhibits adipogenesis by suppressing the transcription of peroxisome proliferator activated receptor gamma 2 (Ppar-γ2) in the nucleus, which is the main facilitator of adipogenesis. However, Arid5a interacts with Il6 mRNA and is exported in the cytoplasm along with UPF-1 via the chromosomal region maintenance 1 (CRM1) pathway in response to inflammation, thereby leading to the mRNA stabilization of not only Il6 but also other inflammatory genes (Tbet, Stat3, etc.) [14]. Nonetheless, the precise molecular mechanism underlying nucleocytoplasmic Arid5a transport needs to be elucidated and understanding this mechanism may further aid in developing strategies to block the nuclear export of Arid5a to reduce inflammation.
IL-17 is known not only to drive immunity against fungal pathogens such as Candida albicans (C. albicans) but also to contribute to autoimmune diseases [15]. Regnase-1, an RNase, constrains the expression of IL-17-induced Il6 and Lcn2 and thus works as a negative regulator in the IL-17 pathway [16]. A group recently investigated whether Arid5a drives the IL-17-induced immune response in mouse mesenchymal cells and in human keratinocytes [17] and whether Arid5a interferes with destabilizing function of Regnase-1. The study demonstrated that Arid5a binds directly to the 3′UTRs of multiple target mRNAs and stabilizes the IL-17-induced mRNA transcripts encoding Il6 and CXC chemokines. Furthermore, Arid5a enhances the translation of IL-17-induced TFs such as C/ebpβ and IκBζ, which further augments the expression of the Lcn2 gene. Additionally, we found that Arid5a is significantly involved in the differentiation of naive T helper (Th) cells into type 17 T helper (Th17) cells in a murine experimental autoimmune encephalomyelitis (EAE) model [9] by controlling the stability of Stat3 mRNA (Figure 1) [10].Arid5a-deficient mice are highly resistant to developing EAE, which is due to a significant decrease in the IL-6 serum level. Interestingly, the number of IL-17-producing CD4+ T cells is critically reduced in Arid5a-/- mice. Arid5a binds with the stem regions of the Il6 and Stat3 3′UTRs via its residue R128, and Regnase-1 binds to these 3′UTRs as well [10]. Therefore, by interfering in binding to the 3′UTR, Arid5a reduces the destabilizing effect of Regnase-1 on these mRNAs and contributes to the development of autoimmune inflammatory diseases. Additionally, Regnase-1 represses the activation and differentiation of CD4+ T cells by destabilizing the mRNAs of Ox40, inducible T-cell costimulator (Icos) and cytotoxic T-lymphocyte-associated protein 4 (Ctla4) [18]. The adoptive transfer of Arid5a-/- encephalitogenic CD4+ T cells ameliorates EAE, which is related to the implication of the Arid5a/Ox40 axis in IL-17 production and EAE development in CD4+ T cells [19]. Moreover, a balance between Arid5a and Regnase-1 is likely to regulate Ox40 mRNA in CD4+ T cells under Th17-polarizing conditions.
Some IL-17-dependent pathways that lead to mRNA stabilization are initiated through IL-17 receptor complex adaptors such as Act1, TRAF2, and TRAF5 [20-22]. A subsequent study reported a role of SEFIR domain of Act1 in mediating Act1 RNA binding activity to stabilize mRNAs of the pro-inflammatory genes (Tnf, Csf2 and Cxcl1) in IL-17-stimulated conditions [23]. In order to control mRNA metabolism, Act1 directly binds to the 3’-UTRs of inflammatory mRNAs and form distinct ribonucleic proteins (RNPs) in processing (P)-bodies. This binding between Act1 and mRNAs can be diminished by RNA aptamers, which results in attenuation of IL-17-mediated mRNA stabilization [23]. However, Act1 does not interact with Arid5a, both could associate with TRAF2 [17]. It is likely that Act1 might regulate Arid5a mRNA under IL- 17 stimulated conditions and both could mediate IL-17- mediated mRNA stabilization and translational control.
The unsolicited production and release of products by immune cells are often the reason for cytokine storm, which is characterized by varying degrees of pathological reactions including vascular expansion, high fever, respiratory disorders, and the secretion of secondary mediators, such as nitric oxide and reactive oxygen species (ROS), resulting in tissue damage [24]. We reported that Arid5a-/- mice are less susceptible to bleomycin (BLM)- induced lung injury and show nominal inflammation or edema in the lung tissue [25]. ROS production in the bronchoalveolar lavage fluid (BALF) is observed to be lower in BLM-treated Arid5a-/- mice, and this reduction is associated with a reduced OxPAPC level in the lung tissue of these mice. Additionally, Arid5a and Il6 gene expression is induced in response to OxPAPC stimulation in murine alveolar macrophages. Further studies are needed to delineate the mechanism of the Arid5amediated regulation of ROS. In addition, sepsis is a worthy example of a severe systemic inflammatory response to infection associated with cytokine storm. We showed that Arid5a-/- T cells are defective in expressing IFN-γ under Th1-polarizing conditions, as T-bet mRNA expression is inhibited [11]. Arid5a is involved in binding with the conserved stem-loop structure of the 3′UTR of Tbet and stabilizing Tbet mRNA. Arid5a-/- mice are also resistant to Propionibacterium acnes-primed LPS injection, an IFN-γ-dependent endotoxic shock model. Overall, Arid5a regulates the augmentation of IFN-γ and IL-6 levels via the stabilization of Tbet and Il6 mRNAs, respectively, which possibly works synergistically to amplify the expression of various other cytokines that ultimately lead to the development of septic shock in mice (Figure 1).
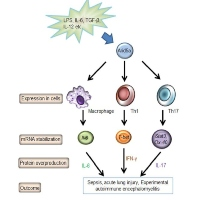
Figure 1. A schematic diagram illustrating Arid5a-mediated mRNA stability, inflammation and autoimmunity.
Ample amount of data has demonstrated that cardiac fibroblasts (CFs) regulate the cardiac function through the secretion of various cytokines and growth factors [26,27]. Angiotensin II-stimulated CFs released IL-6 family proteins and enlarged cardiomyocytes [27]. Thus, one of the promising strategies against cardiac remodeling could be the inhibition of the release of pro-inflammatory cytokines from CFs. It is well-known that β-adrenergic receptor (βAR) agonist enhances the production of proinflammatory cytokines [28,29] which results in the onset and/or progression of heart failure. A recent study has shown a mechanism by which how βAR stimulation induces the production of pro-inflammatory cytokines by using adult murine CFs [30]. The data have suggested that β2-adrenergic stimulation post-transcriptionally upregulates the expression of Il6 by the induction of Arid5a through cAMP/PKA/CREB pathway in adult CFs, which is further directed that β2AR/Arid5a/IL-6 axis could be a therapeutic target against cardiac inflammation [30].
Immunity and inflammation are essential elements of tumor microenvironment which provides a nurturing niche for cancer [31]. A German group has more recently reported the association of Arid5a in lung cancer [32]. It is well reported that tumor-associated macrophages (TAMs) influence lung tumorigenesis by inducing immunosuppression. The functional heterogeneity of TAMs is reflected by phenotypic subsets such as tumorinhibiting M1-like and tumor-promoting M2-like TAMs. However, the precise mechanisms by which tumor microenvironment induces the phenotypic transition of M1-like TAMs to M2-like TAMs are not yet fully understood [33,34]. By transcriptome analysis, Sarode et al. have found upregulation of Wnt/β-catenin signaling in TAMs isolated from human lung tumor. Pharmacological inhibition and macrophage-specific genetic alteration of β-catenin gene reprogrammed M2-like TAMs to M1-like TAMs confirmed in both in vitro and in vivo models. This reprogramming of TAMs was associated with the suppression of primary and metastatic lung tumor growth. The higher expression of β-catenin, and β-catenin-mediated transcriptional activation of FOS-like antigen 2 (FOSL2) and inhibition of the Arid5a are responsible to initiate gene regulatory switch from M1-like TAMs to M2-like TAMs. Furthermore, higher expression of β-catenin and FOSL2 are correlated with poor prognosis of patients with lung cancer. Thus, targeting β-catenin/FOSL2/Arid5a axis in macrophages may offer a new immunotherapeutic option in lung cancer. Collectively, these reports highlight the importance of Arid5a-mediated transcriptional and posttranscriptional regulation of inflammatory mRNAs through its nucleic acid binding specificity and disease outcome. Moreover, a better understanding of Arid5a protein and its interaction with mRNAs may help to improve therapeutic and diagnostic modalities targeting Arid5a.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Funding
This work was supported by the advanced postdoc’s research grant from the Immunology Frontier Research Center, Osaka University and Chugai Pharmaceutical Co. Ltd., Japan.
Acknowledgements
We thank Mari Okawa for her assistance.
References
2. Choy EH, Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol. 2020 Jun; 16(6):335-45.
3. Nyati KK, Agarwal RG, Sharma P, Kishimoto T. Arid5a Regulation and the Roles of Arid5a in the Inflammatory Response and Disease. Front Immunol. 2019 Dec 5;10:2790.
4. Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009 Apr 30;458(7242):1185-90.
5. Turner M, Díaz-Muñoz MD. RNA-binding proteins control gene expression and cell fate in the immune system. Nat Immunol. 2018 Feb;19(2):120-9.
6. Anderson P. Posttranscriptional control of cytokine production. Nat Immunol. 2008 Apr;9(4):353-9.
7. Stoecklin G, Gross B, Ming XF, Moroni C. A novel mechanism of tumor suppression by destabilizing AU-rich growth factor mRNA. Oncogene. 2003 Jun 5;22:3554-61.
8. Amano K, Hata K, Muramatsu S, Wakabayashi M, Takigawa Y, Ono K, et al. Arid5a cooperates with Sox9 to stimulate chondrocyte-specific transcription. Mol Biol Cell. 2011 Apr 15;22(8):1300-11.
9. Masuda K, Ripley B, Nishimura, R, Mino T, Takeuchi O, Shioi G, et al. Arid5a controls IL-6 mRNA stability, which contributes to elevation of IL-6 level in vivo. Proc Natl Acad Sci U.S.A. 2013 Jun 4; 110(23): 9409-14.
10. Masuda K, Ripley B, Nyati KK, Dubey PK, Zaman MM, Hanieh H, et al. Arid5a regulates naive CD4+ T cell fate through selective stabilization of Stat3 mRNA. J Exp Med. 2016 Apr 4; 213(4): 605-19.
11. Zaman MM, Masuda K, Nyati KK, Dubey PK, Ripley B, Wang K, et al. Arid5a exacerbates IFN-?-mediated septic shock by stabilizing T-bet mRNA. Proc Natl Acad Sci U.S.A. 2016 Oct 11; 113(41): 11543-8.
12. Masuda K, Kimura A, Hanieh H, Nguyen NT, Nakahama T, Chinen I, et al. Aryl hydrocarbon receptor negatively regulates LPS-induced IL-6 production through suppression of histamine production in macrophages. Int Immunol. 2011 Oct;23(10):637-45.
13. Chalise JP, Hashimoto S, Parajuli G, Kang S, Singh SK, Gemechu Y, et al. Feedback regulation of Arid5a and Ppar-?2 maintains adipose tissue homeostasis. Proc Natl Acad Sci U.S.A. 2019 Jul 23;116(30):15128-33.
14. Higa M, Oka M, Fujihara Y, Masuda K, Yoneda Y, Kishimoto T. Regulation of inflammatory responses by dynamic subcellular localization of RNA-binding protein Arid5a. Proc Natl Acad Sci U.S.A. 2018 Feb 6;115(6),E1214- 20.
15. Conti HR, Gaffen SL. IL-17-mediated immunity to the opportunistic fungal pathogen Candida albicans. J Immunol. 2015 Aug 1;195(3): 780-8.
16. Garg A, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 endoribonuclease activity negatively regulates interleukin-17-mediated signaling and inflammation. Immunity. 2015 Sep 15;43(3): 475-87.
17. Amatya N, Childs EE, Cruz JA, Aggor FEY, Garg AV, Berman AJ, et al. IL-17 integrates multiple self-reinforcing, feed-forward mechanisms through the RNA binding protein Arid5a. Sci Signal. 2018 Oct 9; 11(551): eaat4617.
18. Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, et al. Regnase-1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. 2015;161(5),1058-73.
19. Hanieh H, Masuda K, Metwally H, Chalise JP, Mohamed M, Nyati KK, Standley DM, Li S, Higa M, Zaman MM, Kishimoto T. Arid5a stabilizes OX40 mRNA in murine CD4+ T cells by recognizing a stem-loop structure in its 3’UTR. Eur J Immunol. 2018 Apr;48(4):593-604.
20. Sønder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U. IL-17-induced NF-kappaB activation via CIKS/Act1: Physiologic significance and signaling mechanisms. J Biol Chem. 2011 Apr 15;286(15):12881-90.
21. Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicingregulatory factor SF2 (ASF2). Nat Immunol. 2012 Aug 7;12(9):853-60.
22. Herjan T, Yao P, Qian W, Li X, Liu C, Bulek K, et al. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J Immunol. 2013 July 15;191(2):640-9.
23. Somma D, Mastrovito P, Grieco M, Lavorgna A, Pignalosa A, Formisano L, Salzano AM, Scaloni A, Pacifico F, Siebenlist U, Leonardi A. CKIS/DDX3X interaction controls the stability of Zc3h12a mRNA induced by IL-17. J Immunol. 2015 Apr 1;194(7): 3286-94.
24. Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 2014 Oct;13(10):741-58.
25. Dubey PK, Masuda K, Nyati KK, Zaman MM, Chalise JP, Millrine D, et al. Arid5a-deficient mice are highly resistant to bleomycin-induced lung injury. Int Immunol. 2017;29(2):79-85.
26. Porter KE, Turner NA. Cardiac fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther. 2009 Aug;123(2):255-78.
27. Sano M, Fukuda K, Kodama H, Pan J, Saito M, Matsuzaki J, et al. Interleukin-6 family of cytokines mediate angiotensin II-induced cardiac hypertrophy in rodent cardiomyocytes. J Biol Chem. 2000 Sep 22;275(38):29717-23.
28. de Lucia C, Eguchi A, Koch WJ. New insights in cardiac beta-adrenergic signaling during heart failure and aging. Front Pharmacol. 2018 Aug 10;9:904.
29. Murray DR, Prabhu SD, Chandrasekar B. Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation. 2000;101(20):2338-41.
30. Tanaka S, Imaeda A, Matsumoto K, Maeda M, Obana M, Fujio Y. ?2-adrenergic stimulation induces interleukin-6 by increasing Arid5a, a stabilizer of mRNA, through cAMP/PKA/CREB pathway in cardiac fibroblasts. Pharmacol Res Perspect. 2020 Apr;8(2):e00590.
31. Mantovani A, Ponzetta A, Inforzato A, Jaillon S. Innate immunity, inflammation and tumour progression: doubleedged swords. J Intern Med. 2019 May;285(5):524-32.
32. Sarode P, Zheng X, Giotopoulou GA, Weigert A, Kuenne C, Günther S, et al. Reprogramming of tumorassociated macrophages by targeting β-catenin/FOSL2/ ARID5A signaling: A potential treatment of lung cancer. Sci Adv. 2020 Jun 5;6(23):eaaz6105.
33. Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J Immunol. 2005 Jul 1;175(1):342-9.
34. Zheng X, Turkowski K, Mora J, Brüne B, Seeger W, Weigert A, et al. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget. 2017 Jul 18;8(29):48436-52.