Abstract
Background: Recurrent infections in children may indicate underlying inborn errors of immunity. Early detection by combined immunologic and genetic testing is essential for accurate diagnosis and targeted intervention.
Methods: This prospective study included 60 pediatric patients aged ≤18 years with recurrent infections, evaluated between 2022 and 2024. The median age at enrollment was 6.2 years ([IQR] 3.6–11.3). Immunological assessment included flow cytometric immunophenotyping of lymphocyte subsets, serum immunoglobulin profiling (IgG, IgA, IgM, IgE and IgG subclasses), complement (C3, C4), and vitamin D measurement. Microbiological cultures of sputum or pharyngeal swabs were collected. Whole Exome Sequencing (WES) was performed for genetic evaluation and results verified by Sanger sequencing.
Results: A subset of patients had reduced CD4+ T cells, memory B cells or NK cells. Hypogammaglobulinemia, particularly of IgG2 and IgA subclasses, was common. Pathogenic bacteria, mainly Haemophilus influenzae and Staphylococcus aureus, were isolated in 45% of cases. WES identified pathogenic or likely pathogenic variants in TNFRSF13B, CFTR, IL10RA, MEFV and other genes. A possible association between MBL2 c.161 G>A and altered CD4+ T cell percentages, lower NK cell numbers and reduced CD8+ effector T cells could indicate immunomodulatory effects.
Conclusion: Comprehensive immunogenetic profiling reveals distinct patterns of immune dysregulation and genetic susceptibility, supporting personalized diagnostic and therapeutic strategies, including immunoglobulin substitution, immunomodulation, and genetic counseling.
Keywords
Recurrent infections, Inborn errors of immunity, Immunophenotyping, Whole-exome sequencing, SNP analysis
Introduction
Recurrent infections in children represent a major clinical challenge and often lead to frequent hospitalizations, prolonged antibiotic use and increased morbidity. While occasional infections are common in childhood, an unusually high frequency or severity of infections may indicate an underlying immunologic or genetic predisposition. Ten percent of children with recurrent infections have an immunodeficiency, i.e. a defect in one or more components of the immune system [1]. Inborn errors of immunity (IEIs), formerly called primary immunodeficiency disorders (PIDs) encompass a group of more than 550 inherited disorders, often due to single-gene mutations, that result in the specific impairment of normal immune development and function [2]. Some of patients with IEI do not initially present with recurrent infections, but with features of immune dysregulation that includes severe atopy, autoinflammatory or autoimmune diseases, lymphoproliferation and increased risk of malignancies [3]. Accurate diagnosis is essential for appropriate patient management, including immunologic and genetic profiling.
The initial evaluation of a child suspected of having an inborn error of immunity (IEI) includes a detailed medical history, physical examination and targeted immunologic testing [1]. A complete blood count (CBC) should be checked for lymphopenia (indicative of T-cell deficiency) and thrombocytosis (indicative of chronic inflammation). Elevated ESR and/or CRP may indicate chronic infection or autoimmunity. Conversely, the absence of fever or an increase in CRP in systemic infections may indicate an immune disorder.
Immunoglobulin levels must be age-adjusted as they change with development. Low values indicate antibody or combined immunodeficiencies. Elevated IgE levels can indicate allergies, eczema or hyper-IgE syndromes; extremely high IgE levels can occur in monogenic atopy or hyper-IgE immunodeficiencies [4]. Functional antibody defects can also occur with normal IgG. Severe reductions in all immunoglobulin classes and B cells are indicative of congenital agammaglobulinemia.
Flow cytometry for T cells (CD3), B cells (CD19) and NK cells (CD56) is an important early test. A significantly reduced number of naïve T cells–with or without reduced B/NK cells–is a hallmark of severe combined immunodeficiency (SCID). Counting naïve T cells also helps to assess thymus development.
Conventional diagnostic tests may not be conclusive. For example, antibody production may be impaired despite normal immunoglobulin levels, making antibody deficiency difficult to detect. Autoantibody-based tests can give false-negative results in hypogammaglobulinemia, as is the case in common variable immunodeficiency (CVID) or X-linked agammaglobulinemia (XLA) [5]. Therefore, genetic testing is often required for an accurate diagnosis. Advanced genomic technologies such as Whole Exome Sequencing (WES) offer a comprehensive approach for identifying rare and novel genetic variations associated with immune dysfunction. It has proven to be highly effective in diagnosing monogenic disorders, including rare immune-related genetic defects, but also can provide insights into polygenic risk factors and susceptibility loci associated with immune dysregulation. By integrating WES and SNP profiling, researchers are able to identify both rare pathogenic variants and broader genetic predispositions, leading to a more comprehensive understanding of recurrent infections in children [6].
Our study aims to investigate the genetic and immunologic factors contributing to recurrent infections in children by integrating a comprehensive immunological workup and whole-exome sequencing analysis.
Materials and Methods
Study population
Sixty children (≤18 years; 32 males and 28 females) with recurrent infections were recruited between 2022 and 2024. The median age at enrollment was 6.2 years (interquartile range [IQR] 3.6–11.3). To account for age-related differences in immune system maturation, participants were stratified into three predefined age groups: 1–5 years, 6–11 years, and 12–18 years. Sex was recorded for all participants and included in the descriptive analyses.
Inclusion criteria
- Recurrent infections with suspected primary immunodeficiency
- Severe allergic reactions and frequent respiratory tract infections
- Clinically and laboratory-confirmed immunodeficiency
Exclusion criteria
- Genetically confirmed cystic fibrosis
- Ongoing chemotherapy or immunosuppressive therapy
- Active tuberculosis or other severe systemic infections at enrollment (eligible ≥6 months post-treatment)
Only patients with written parental informed consent were included as participants. The study was approved by the institutional ethics committee (Figure 1).
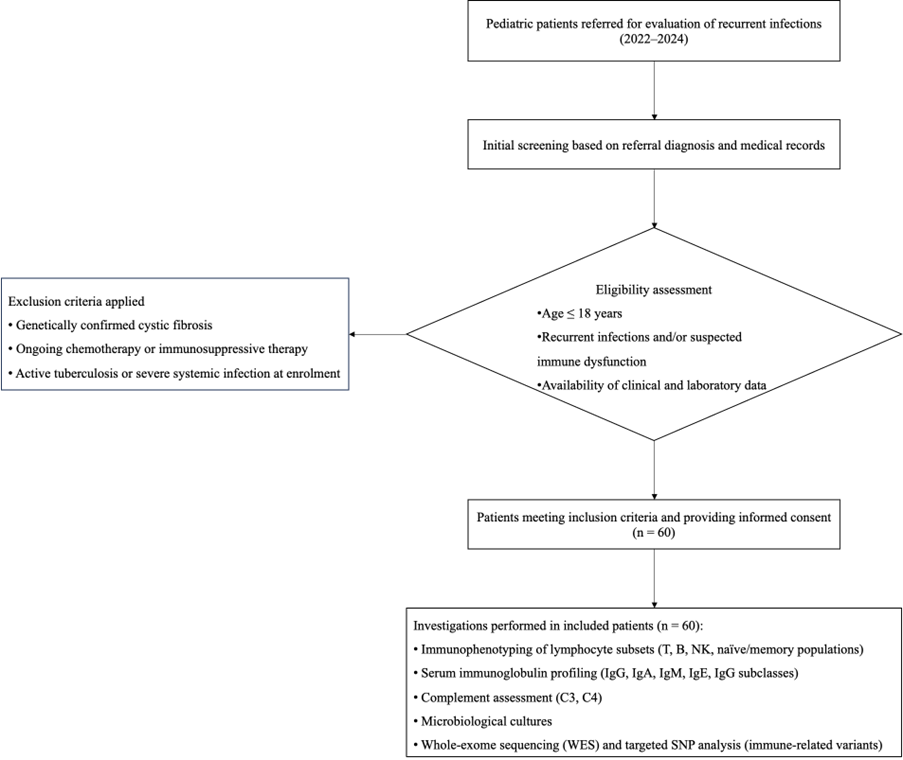
Figure 1. Flowchart of patient enrolment, screening, eligibility assessment, exclusion criteria, and final inclusion in the study cohort.
Quantitative data were derived from a structured expert review of hospital medical records; values reflect documented clinical findings at the time of assessment.
Laboratory testing
Immunological examination: Quantification of serum IgM, IgG, IgA, IgG subclasses (IgG1–IgG4), IgE and complement components C3 and C4 was performed by turbidimetry using OptiliteR Protein Analyzer and commercial OptiliteR kits for the listed analytes. Immunophenotyping of lymphocytes in peripheral blood was performed by multi-color flow cytometric method. Monoclonal antibodies conjugated to different fluorochromes was used to determine the following immune cell subsets: T lymphocytes (CD3+), activated T lymphocytes (CD3+HLA-DR+), helper-inducer T lymphocytes (CD3+CD4+), suppressor-cytotoxic T lymphocytes (CD3+CD8+), naïve (CD45RA+CD62L+), central memory (CD45RA-CD62L+), effector memory (CD45RA-CD62L-), terminally differentiated effectors (CD45RA+CD62L-) CD4+ and CD8+ T lymphocytes, B-lymphocytes (CD19+), naïve (IgD+CD27-), non class switched memory (IgD+CD27+), class switched memory (IgD-CD27+) B cells, NK cells (CD3-CD16&56+), NKT cells (CD3+CD16&56+), CD8+CD11b+ cells, CD8+CD11b- cells, Treg (CD4+CD25+CD127low), Th17 (CD4+CD161+CD196+) cells. Briefly, 50 μl aliquots of blood (anticoagulant sodium heparin) were incubated in BD Falcon 12x75mm tubes for 20 minutes in the dark at room temperature with combinations of optimally titrated fluorochrome-conjugated monoclonal antibodies. After surface staining, erythrocytes were lysed for 10 minutes with 1 ml BD FACSLysing solution diluted according to the manufacturer's instructions. After lysis, the cell suspension was centrifuged at 600 g for 5 minutes, the supernatant was removed and 1 ml CELL WASH buffer (Becton Dickinson) was added to the cells. The labeled immune cells were washed by centrifugation at 600 g for 5 minutes. The supernatant was removed and the cells were resuspended in 350 μl of CELL FIX (Becton Dickinson). The following monoclonal antibodies (manufactured by BD Pharmingen™, Becton Dickinson and Thermo Fisher Scientific ) and panel combinations (manufactured by BD Pharmingen™, Becton Dickinson) were used in order to distinguish the variety of cell subsets listed above: CD3 FITC/CD16&56 PE/CD45 PerCP/CD19 APC CE-IVD BD Multitest™; CD3 FITC/CD8 PE/CD45PerCP/CD4 APC BD Multitest™; CD45RA FITC/CD62L PE/CD3 PerCP/CD8 APC BD Multitest™; CD45RA FITC/CD62L PE/CD3 PerCP/CD4 APC BD Multitest™; CD4 FITC/CD25 PE/CD 3 PerCP/CD 127 APC; CD4 FITC/CD1 161 PE/ CD3 PerCP/CCR6PeCys; BD CD57 FITC/BD 8 APC; CD8 FITC/CD11b PE (Thermo Fisher Scientific); CD4 FITC/ CD161 PE/ CD 196 PerCP-Cy/ (BD Pharmingen™, Becton Dickinson).
Sample collection and analysis were performed on a FACS Canto II flow cytometer, using FACSDiva v6.2 software (Becton Dickinson). At least 10,000 events were collected from each sample in the lymphocyte gate. Initial assessment of the viable portion of the sample was performed by physical parameter selection based on forward (FSC) and side (SSC) scatter, with the lymphocyte population identified by low forward and side scatter and checked for purity by CD45 positivity. The different lymphocyte subpopulations were then identified by immune phenotype markers. Total leukocyte counts were measured with a hematology analyzer. The absolute number of each lymphocyte subpopulation was calculated by multiplying the percentage of the lymphocyte subpopulation by the absolute lymphocyte count.
The results were compared with the reference values for the population-based respective age group of the patients and classified as low, normal and high for better visualization in the diagrams.
Immunological sampling was conducted during periods of clinical stability. All children were afebrile, showed no signs or symptoms of acute infection at the time of blood collection, and were not receiving antimicrobial or antiviral therapy. Although the exact interval since the last infectious episode was not consistently recorded due to the retrospective design, samples were collected outside clinically apparent acute infections to reflect baseline immune status rather than transient changes driven by infection.
Microbiological examination
Upon admission, sputum or pharyngeal swabs were collected and transported in standard medium. Samples were cultured at 37°C for 24–48 hours using blood agar, MacConkey agar, Candida chromium agar, and selective chocolate agar with vancomycin for Moraxella and Haemophilus spp., as per in-house protocols. Cultures were incubated under aerobic or microaerophilic conditions. Isolates were identified using Crystal BD BBL and Remel RapID systems. A bacterial load >105 CFU/mL was considered clinically significant. Antimicrobial susceptibility was assessed via the Kirby-Bauer disk diffusion method, with MIC values interpreted according to CLSI standards.
Serum 25(OH)-vitamin D levels were quantified using LC-MS/MS with triple quadrupole detection. The method was validated per clinical and industrial standards and certified via DEQAS (UK).
Genetic testing
Genomic DNA from the patients included in this study was extracted from K2EDA blood samples by standard salt extraction methodology. Parents’ DNA samples were also available for segregation analysis when needed.
Whole Exome Sequencing (WES) analysis was performed using Illumina sequencing-by-synthesis technology. It was conducted in the partner laboratories "Admera Health, LLC", USA and xGen Exome Research Panel (IDT, Integrated DNA Technologies, Inc. USA) whole exome capture-based method was used. Sequences were aligned to the human genome reference sequence (GRCh37 / hg19) for variant calling and annotation.
A panel of 336 immune deficiency-related-genes was analyzed for each patient exome (Supplementary Table 1) via a specialized software GensearchNGS, (PhenoSystems SA, Switzerland).
Additionally, 17 target polymorphic regions, associated with immune response modification (presented in Supplementary Table 2) were analyzed for single nucleotide polymorphism (SNP) genotype profiling. SNP positions and nomenclature followed the latest dbSNP and Ensembl databases [7].
The genetic variants of interest were confirmed and segregated in the families by Sanger sequencing with BigDye® Terminator v3.1 kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s protocols. ABIPrism 3130 Genetic Analyzer was used for electrophoretic separation and data received was processed with Sequencing Analysis v5.1.1 software.
The interpretation of the detected genetic variants was performed according to the classification criteria of the guidelines of the American College of Medical Genetics and Genomics/Association of Molecular Pathology (ACMG/AMP), taking into account the clinical manifestations and the results from the segregation analyses, performed in the families. Variant positions and nomenclature followed the latest dbSNP and Ensembl databases.
Statistical analysis
All statistical analyses were performed with IBM SPSS Statistics Version 26 (IBM Corp., USA). Descriptive statistics were used to summarize the data. Normality of continuous variables was tested using the Shapiro-Wilk test. For comparisons between several groups, a one-way analysis of variance (ANOVA) followed by a Dunnett post-hoc test was used for normally distributed data, while the Kruskal-Wallis H test followed by a pairwise Mann-Whitney U test was used for non-normally distributed data. For categorical variables, group associations were analyzed using the chi-square test or Fisher’s exact test if the expected frequencies were low. Age- and sex-adjusted analyses were performed using generalized linear models with normal distribution and identity link where statistical significance was assessed using Type III tests of model effects. A p-value <0.05 was considered statistically significant in all analyses.
Results
Demographic characteristics
The cohort comprised 27 children aged 1–5 years (45.0%), 18 children aged 6–11 years (30.0%), and 15 adolescents aged 12–18 years (25.0%).
Regarding sex distribution, 32 patients were male (53.3%) and 28 were female (46.7%), resulting in a relatively balanced male-to-female ratio.
Clinical data
Clinical severity and exposure data were obtained through expert review of comprehensive hospital medical records. Selected baseline characteristics are summarized in Supplementary Table 1. Recurrent respiratory infections were documented in all patients. A history of pneumonia was reported in 22 patients (37%), while frequent antibiotic exposure was recorded in 42 patients (70%). Hospitalizations due to infectious complications occurred in 25 patients (42%). Recurrent wheezing or broncho-obstructive episodes were observed in 27 patients (45%), most commonly with onset in early childhood. Regarding immunization status, 49 patients (82%) were regularly vaccinated according to the national immunization schedule, whereas delayed or irregular vaccination was documented in 11 patients (18%). Physician-diagnosed asthma was present in 16 patients (27%), including only children with a clearly established diagnosis of bronchial asthma or those receiving long-term controller therapy with inhaled corticosteroids, with or without leukotriene receptor antagonists. Concomitant allergic rhinitis was identified in 9 patients (15%) and included allergic rhinitis, rhinoconjunctivitis, or chronic allergic rhinosinusitis; isolated viral rhinitis or non-specific nasal symptoms without an allergic context were excluded. Atopic dermatitis was recorded in 12 patients (20%). Food allergies were documented in 15 patients (25%), most commonly to cow’s milk protein (9 children), followed by nut allergies (6 children). Documented drug hypersensitivity reactions were reported in 10 patients (17%), most frequently to β-lactam antibiotics or non-steroidal anti-inflammatory drugs. Autoimmune cytopenias were identified in 14 patients (23%), most commonly idiopathic thrombocytopenia (12 patients, 20% of the cohort), followed by neutropenia (1 patient) and agranulocytosis (1 patient). Chronic spontaneous urticaria was noted in 7 patients (12%). Arthritis was recorded in 3 patients, and alopecia in 1 patient. Clinically suspected autoinflammatory conditions were uncommon, affecting 5 patients (8%), and included only cases with PFAPA-like phenotypes without a definitive diagnosis.
Immunological results
The analysis of the data includes both the absolute number (cells/µl) and the relative proportion of the different immune cell populations, which provides a deeper understanding of immune regulation and balance.
In the analyzed patient cohort, the absolute number of T lymphocytes (CD3+) ranges between 650 and 3250 cells/µl, with a mean of 1785.2 cells/µl, while their percentage varies between 50% and 86%, with an average of 66.1% (Figure 2). This indicates a relatively well-preserved T cell compartment, although variations within the population could indicate individual variations in immunocompetence. Activated T lymphocytes (CD3+DR+) show a wide range in both absolute number (26 to 1848 cells/µl, mean 195.7 cells/µl) and percentage (1% to 56%, mean 7.3%), indicating fluctuations in immune activation that could be related to ongoing or overcome infections. The absolute numbers of helper T cells (CD3+CD4+) ranged from 364 to 1700 cells/µL, with a mean of 875.1 cells/µL, while their percentage ranged from 17% to 47%, averaging 32.6%. Naïve CD4+ T lymphocytes (CD45RA+CD62L+) have a mean percentage of 54.2% and a wide range (5.2% to 89.1%), indicating variations in immune maturation and response. CD4+ T cells with central memory (CD45RA-CD62L+) and CD4+ T cells with effector memory (CD45RA-CD62L-) have a mean of 29.8% and 12.9%, respectively, consistent with expected immune memory function, although the wide range suggests individual differences in adaptive immunity. Effector CD4+ T cells (CD45RA+CD62L-) are present to a lesser extent, with a mean of 3.0%. Their low frequency likely reflects immune homeostasis in the absence of acute infection, rather than impaired effector differentiation. Regulatory CD4+ T lymphocytes (Tregs, CD25+CD127LOW) make up a smaller proportion of the immune system, averaging 6.7%, and play a crucial role in immune tolerance and regulation. Cytotoxic T lymphocytes (CD3+CD8+) have a mean absolute number of 727.9 cells/µl and a mean percentage of 26.9 %, indicating a relatively stable CD8+ compartment. However, the distribution of naïve, central memory, effector memory and effector CD8+ T cells varies considerably. Naïve CD8+ T cells (CD45RA+CD62L+) predominate with an average percentage of 47.3%, indicating an immune system that still has a reserve of unprimed cytotoxic cells. Central memory CD8+ T cells (CD45RA-CD62L+) have a lower average percentage (11.1 %), while effector memory CD8+ T cells (CD45RA-CD62L-) have an average percentage of 22.3 %, indicating an active role in the immune response. Effector CD8+ T cells (CD45RA+CD62L-) averaged 18.8%, suggesting that a significant portion of the CD8+ pool is capable of immediate cytotoxic function, while the remainder maintains immunological memory.
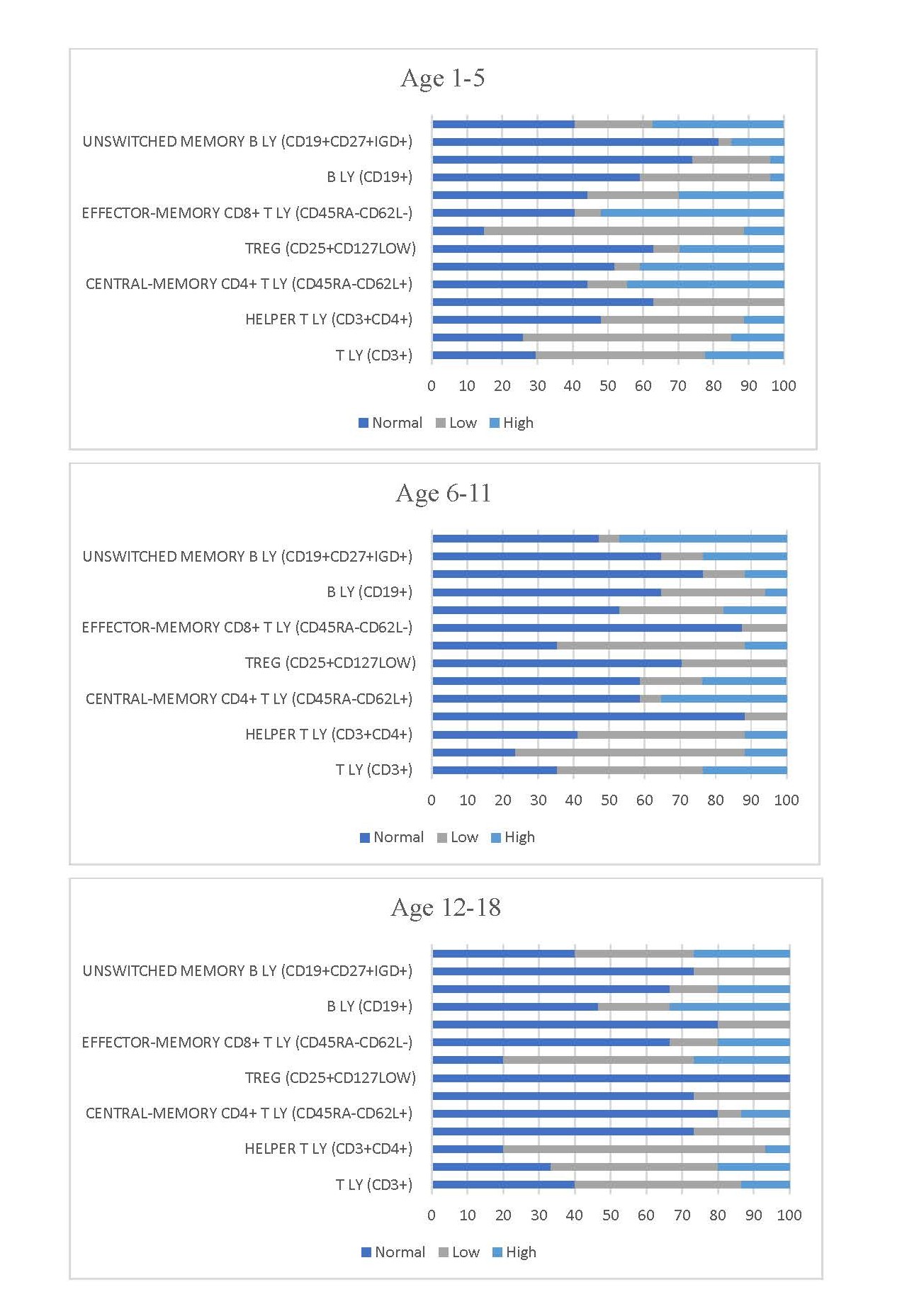
Figure 2. Immunophenotyping profile of lymphocyte subsets. Stacked bars showing distributions of CD4+, CD8+, B, NK, and Treg cells, including naïve, memory, and effector subpopulations in children with recurrent infections.
The absolute number of B lymphocytes (CD19+) ranges between 62 and 1272 cells/µl and averages 429.5 cells/µl, while their proportion varies between 2.5% and 30% and averages 15.7%. Within the B cell population, naïve B cells (CD19+CD27-IGD+) dominate with a mean percentage of 68.3%, while non-switched memory B cells (CD19+CD27+IGD+) and switched memory B cells (CD19+CD27+IGD-) account for 11.9% and 12.0%, respectively. The predominance of naïve B cells may reflect either limited antigen exposure (age-dependent) or impaired B cell maturation, particularly in individuals with recurrent infections.
Natural killer (NK) cells (CD3-CD16&56+) have an absolute number of 78 to 1150 cells/µl, with a mean of 415.3 cells/µl. Their percentage share fluctuates between 3% and 33% and averages 15.9%. The NKT cells (CD3+CD16&56+) show a wide range (9 to 1535 cells/µl) and a mean value of 139.0 cells/µl, indicating individual differences in their contribution to immune surveillance. The CD57+CD8- and CD57+CD8+ T/NK populations also show considerable variation with mean values of 4.2% and 6.7%, respectively, suggesting heterogeneity in immunosenescence and effector function.
Immunoglobulin levels are divided into the categories "low"," "normal" and "high" for different Ig classes and subclasses (Figure 3). The results provide information about possible immunodeficiencies or dysregulations in the population studied.
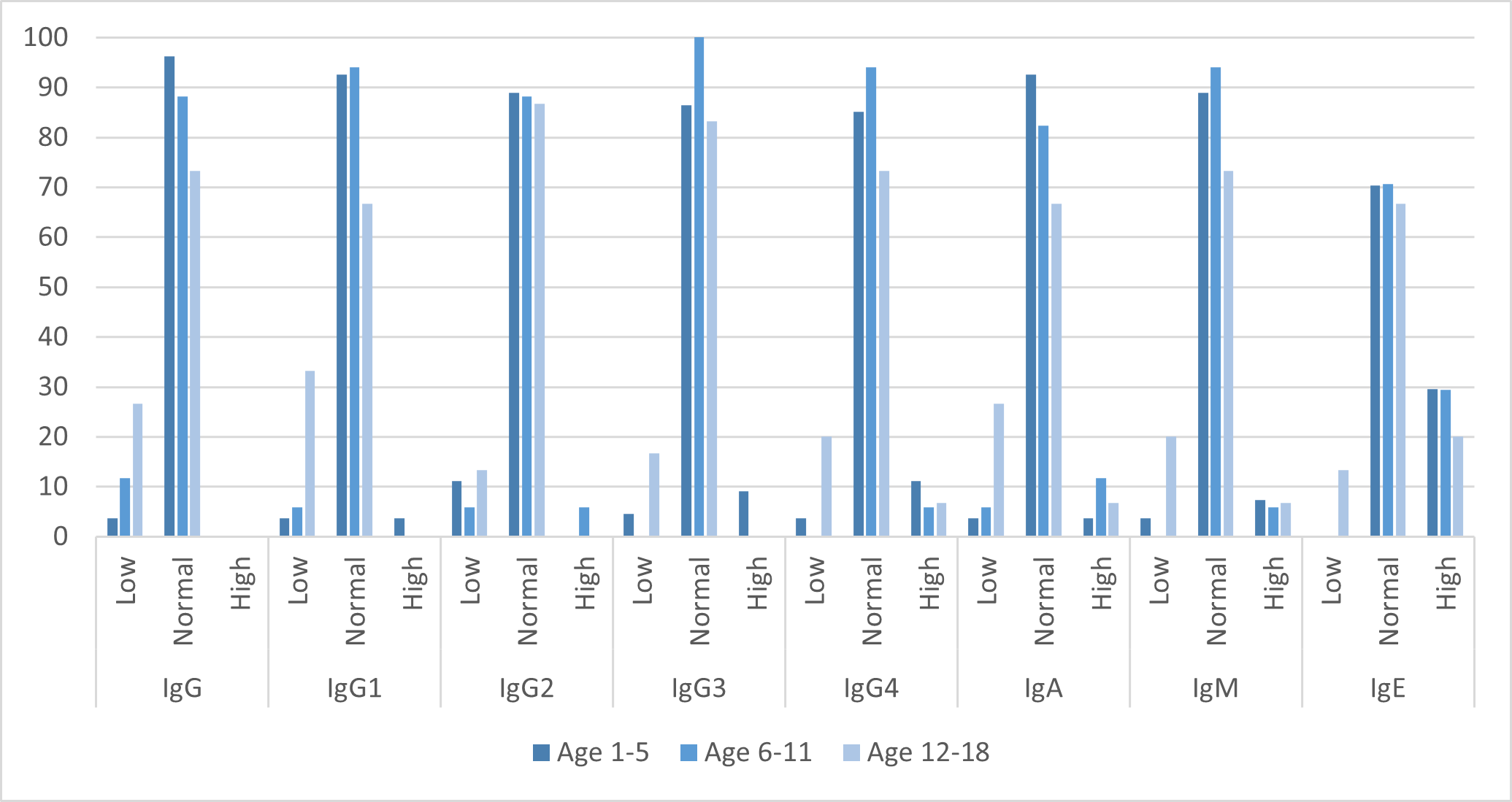
Figure 3. Serum Immunoglobulin Levels in the study cohort. Proportions of patients with low, normal, and elevated levels of total IgG, IgA, IgM, and IgE, as well as IgG subclasses.
Alterations in serum immunoglobulin levels were observed across the study cohort, with a subset of patients demonstrating reduced concentrations of total IgG and IgG subclasses. In particular, decreased levels of IgG1, IgG2, and IgG3 were identified, suggesting impaired humoral immune responses in affected individuals. Deficiencies in these subclasses are clinically relevant, as IgG2 and IgG3 play key roles in immune defense against bacterial pathogens, including encapsulated organisms, and their reduction may contribute to increased susceptibility to recurrent infections.
Reduced IgG4 levels were also noted in a proportion of patients. Although isolated IgG4 deficiency is generally considered of limited clinical significance, it may acquire relevance in the context of recurrent infections or when present alongside other immunological abnormalities.
Alterations in IgA levels were identified in some individuals, raising the possibility of selective IgA deficiency or broader immune dysregulation. Given the central role of IgA in mucosal immunity, particularly within the respiratory and gastrointestinal tracts, reduced IgA levels may further predispose affected patients to recurrent mucosal infections.
Additionally, decreased IgM levels were observed in a subset of patients. As IgM represents the first antibody produced during primary immune responses, its reduction may impair early pathogen recognition and clearance, potentially contributing to recurrent or severe infections. Collectively, these findings indicate the presence of humoral immune abnormalities in a proportion of the study population, which may partially explain the observed pattern of recurrent infections.
IgD levels, although less frequently analyzed in clinical practice, appear to be normal to low in distribution, which is consistent with its limited expression in mature B cells and less clearly defined clinical significance. IgE levels, on the other hand, vary widely, with some individuals having significantly high levels. Elevated IgE levels are often associated with allergic diseases, parasitic infections or hyper-IgE syndromes, while low IgE levels may indicate immune system dysregulation.
C4 levels displayed a non-uniform distribution across low, normal, and elevated ranges. Low C4 levels are often associated with autoimmune diseases such as lupus or hereditary complement deficiencies, which can lead to recurrent infections or immune system dysregulation.
C3 levels also showed a slightly different distribution. Normal C3 levels were most common, followed by low levels, with high levels being the least common. Low C3 levels can occur in autoimmune diseases, chronic infections and complement depletion due to immune activation, while elevated C3 levels can sometimes be associated with inflammatory or metabolic diseases.
Microbiological results
The microbiological results illustrate the distribution of bacterial pathogens identified in the population studied (Figure 4). A significant proportion of cases (55%) had no detectable bacterial growth, which could be due to viral etiologies, organisms that are difficult/non-culturable or suppression due to recent antibiotic therapy. Among the pathogens detected, Haemophilus influenzae was the most frequently isolated bacterium, found in 23.4% of cases, albeit alone in 13% and in combination (predominantly with S. aureus) in the remaining cases. H. influenzae is a common cause of respiratory infections, including sinusitis, otitis media, and pneumonia, especially in individuals with underlying immunodeficiency.
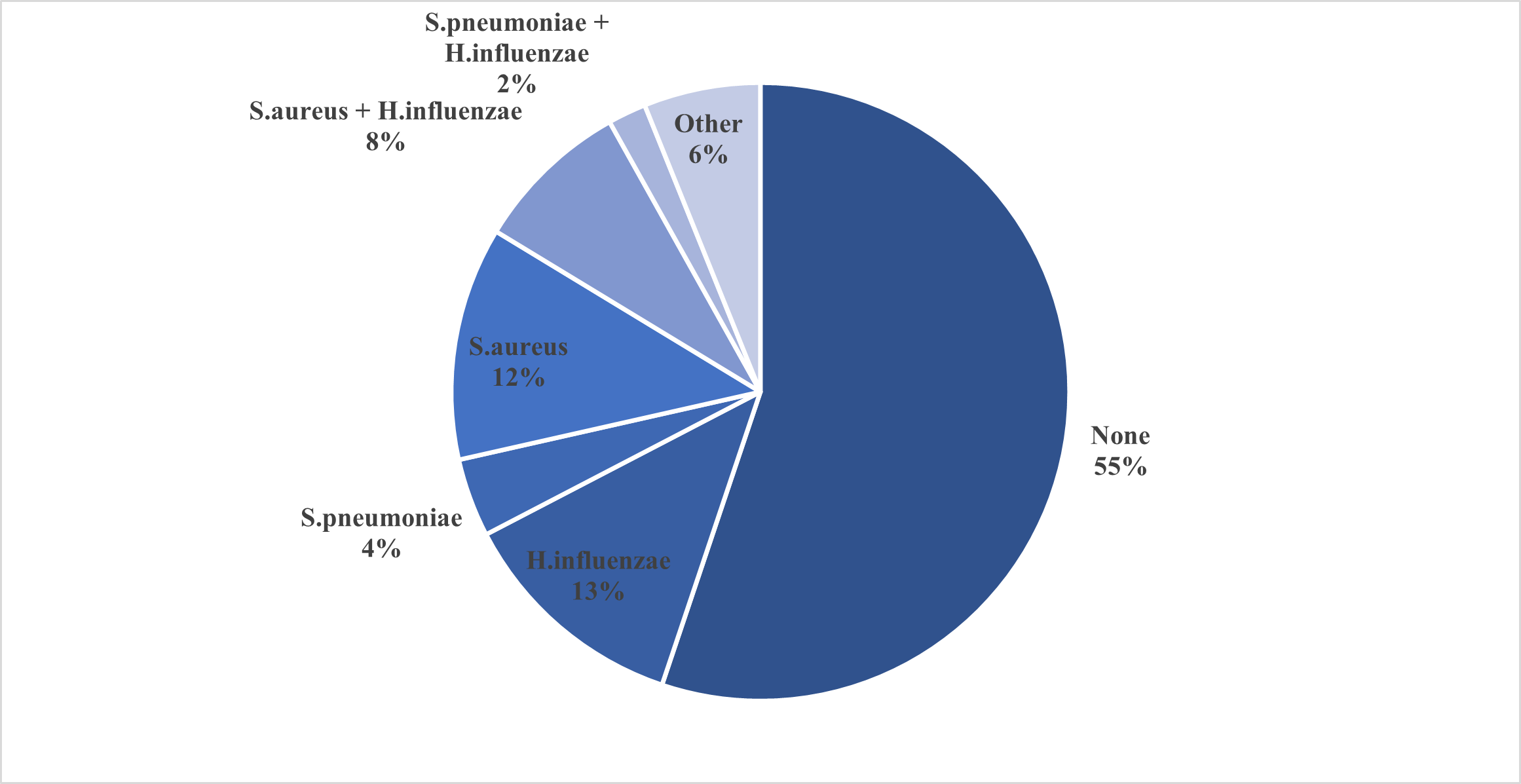
Figure 4. Microbial spectrum of respiratory pathogens. Distribution of cultured pathogens, with H. influenzae and S. aureus being the most frequently isolated organisms.
Staphylococcus aureus was detected in 20.4% of children (but only in 12% as a single pathogen), making it the second most common single organism. This pathogen is known to cause skin and soft tissue infections but can also contribute to more serious illnesses such as pneumonia or bloodstream infections, especially in immunocompromised individuals. Streptococcus pneumoniae, another important respiratory pathogen, was detected in 6% of cases (4% as a single pathogen). Although less common in this dataset, S. pneumoniae remains an important cause of bacterial pneumonia, meningitis and otitis media, particularly in immunocompromised individuals.
Polymicrobial infections have also been observed, with co-infection with S. aureus and H. influenzae occurring in 8% of cases and co-infection with S. pneumoniae and H. influenzae in 2%. These mixed infections may indicate a more severe or chronic disease process, as multiple pathogens can complicate treatment and prolong the disease. In addition, 6% of cases were classified as "other" which were less common bacterial isolates or potentially unidentified organisms.
Laboratory results
In terms of vitamin D levels, a significant proportion of individuals had low vitamin D levels, which is a common finding and may indicate inadequate sun exposure, inadequate diet or underlying metabolic disease. Normal vitamin D levels were observed in a moderate percentage of cases, while high levels were less common. Given the crucial role of vitamin D in immune function, bone health and the regulation of inflammation, these results emphasize the importance of monitoring and supplementing vitamin D as needed.
Genetic results
Comprehensive analysis of 336 different genes was performed; the results from the WES analysis combined with the clinical presentation are presented on Supplementary Table 1. In general, the reference/major alleles were marked as wild-type (WT), and the detected genetic variants were classified according to ACMG/AMP as Benign, Likely-Benign, Variants of uncertain significance (VUS), Likely-Pathogenic and Pathogenic (Figure 5). Ten of the investigated patients were negative for any Pathogenic variants, Likely-Pathogenic variants or VUS. The additional findings including heterozygous carrier status of pathogenic/likely-pathogenic/VUS variants associated with autosomal-recessive conditions were also noted.
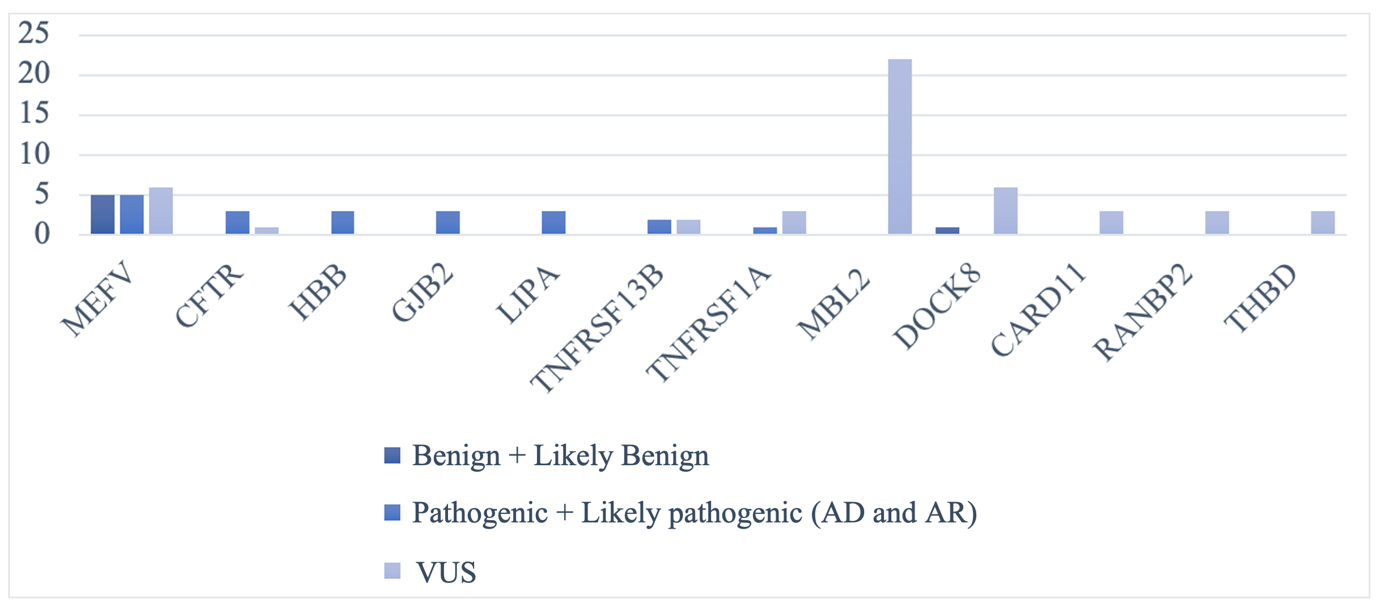
Figure 5. Classification of genetic variants detected by whole-exome sequencing. Most variants detected were classified as variants of uncertain significance (VUS), with a smaller number classified as Pathogenic or Likely-pathogenic.
MEFV gene (which is associated with Familial Mediterranean fever, AD/AR) exhibited a marked diversity. A total of 5 patients were heterozygous for a Pathogenic/Likely-Pathogenic variant (1 for c.2177T>C, p.Val726Ala, 2 - for c.2080A>G, p.Met694Val and 2 – for c.2084A>G, p.Lys695Arg, respectively), thus verifying the possible clinical diagnosis of these children. Six of the patients were heterozygous for variants of uncertain significance: 4 – for c.442G>C, p.Glu148Gln, 1 – for c.910G>A, p.Gly304Arg and 1 – for c.1690T>G, p.Ser564Ala. Five of the patients were heterozygous for the variant c.605G>A, p.Arg202Gln, classified as Benign.
NOD2 results show slight variation with 3 patients carrying the heterozygous variant c.3019dup, p.Leu1007ProfsTer2 categorized as Likely-Benign, 2 patients – with 2 other Benign variants (c.2104C>T, p.Arg702Trp and c.2722G>C, p.Gly908Arg) and 1 patient – with c.922C>T, p.Leu308Phe, classified as VUS.
Interestingly, of the genes examined in the MBL2 no known pathogenic/likely-pathogenic variants were detected, but exclusively 2 VUS were found in the patients studied: c.154C>T, p.Arg52Cys was detected heterozygous in 7 patients; variant c.161G>A, p.Gly54Asp was detected heterozygous in 11 of the patients and homozygous in 1 patient; compound heterozygous for both MBL2 variants described above were 2 of our patients.
Another interesting example for a heterogeneous gene was DOCK8 - with presence of 5 heterozygous variants in the VUS category (1 of these patients was a compound heterozygous carrier of 2 variants).
Several genes showed an occasional presence of Pathogenic or Likely-pathogenic variants, although the majority of our patients remained negative for variants. In particular, 4 of our patients carried TNFRSF13B Pathogenic variants (2 patients were heterozygous for c.310T>C, p.Cys104Arg) or VUS, while 4 patients were heterozygous for 2 VUS in TNFRSF1A (3 were heterozygous for c.362G>A, p.Arg121Gln and one – for c.224C>T, p.Pro75Leu).
Interestingly, CFTR and HBB, both of which expected to be with high carrier frequency among Bulgarian population, demonstrated some presence of Pathogenic variants. Four heterozygous CFTR variants were detected: c.2756A>G, p.Tyr919Cys (Pathogenic/VUS, with reported Bulgarian population frequency in VARSOME database: 0.000375 ), c.3205G>A, p.Gly1069Arg (Likely-Pathogenic/Conflicting interpretation, with reported Global minor allele frequency GMAF in ClinVar database: 0.00040), c.224G>A, p.Arg75Gln (with Conflicting interpretation and reported Bulgarian population frequency in VARSOME database: 0.0146) and c.3983T>C, p.Ile1328Thr (VUS, with reported Global minor allele frequency GMAF in ClinVar database: 0.00020).
According to HBB gene, which is associated with beta-thalassemia minor (heterozygous) or major (homozygous/compound heterozygous), 3 of our patients were heterozygous for the causative mutations c.118C>T, p.Gln40Ter (Pathogenic, known as bo39C/T, with reported frequency among Bulgarians in VARSOME database: 0.00187), c.20A>T, p.Glu7Val (Pathogenic, associated with HbS/Sickle-Cell anemia, with reported Global minor allele frequency GMAF in ClinVar database: 0.02736) and c.-137C>G (Likely-Pathogenic, known also as -87C/G, with reported GMAF in ClinVar database: 0.00020).
Some genes exhibited slightly higher presence of Pathogenic variants – two patients were heterozygous for a Pathogenic variant in FLG, associated with Ichthyosis vulgaris (AR) and Dermatitis, atopic, susceptibility to, 2 (AD). Another two patients were compound heterozygous for two Pathogenic/Likely-Pathogenic variants – each in SPINK5 (associated with Netherton syndrome, AR) and SLC5A2 (associated with Renal glucosuria, AD/AR) respectively. One patient was compound heterozygous for the two most common pathogenic variants in HFE gene associated with Hemochromatosis, type 1 (AR): c.187C>G, p.His63Asp and c.845G>A, p.Cys282Tyr. DNMT3B and ALOXE3, also presented with two patients each compound heterozygous for two variants in the Likely-Pathogenic category, with no other variations, associated with Immunodeficiency-centromeric instability-facial anomalies syndrome 1 (AR) and Ichthyosis, congenital, autosomal recessive 3 (AR), respectively.
In addition, genes such as PIK3CD, STAT1, TLR3 and IL10RA appeared exclusively in the VUS or reference allele categories, with no presence of variants of clear pathogenic classification.
Overall, the dataset shows a strong dominance of wild-type (reference) alleles for all genes, with pathogenic variants occurring only occasionally. However, the presence of numerous VUS suggests a significant number of genetic variations of uncertain clinical significance and emphasizes the need for further research to determine their potential impact.
Analysis of immunological parameters in relation to a targeted MBL2 gene variation c.161 G>A revealed significant differences in both adaptive and innate immune compartments (Figure 6). Individuals carrying the homozygous A/A genotype for the MBL2 c.161 G>A variant exhibited a significantly higher proportion of CD4+ T lymphocytes compared to those with the G/G genotype (p = 0.043), suggesting a potential shift in T cell homeostasis or activation status. In contrast, the absolute number of CD3−CD16+CD56+ NK cells was markedly reduced in the A/A group relative to C/C carriers (p=0.040). Furthermore, subjects harboring variants of uncertain significance (VUS) in MBL2 showed significantly lower frequencies of effector CD8+ T cells (CD8+CD45RA+CD62L−) compared to those with wild-type alleles (p=0.023).
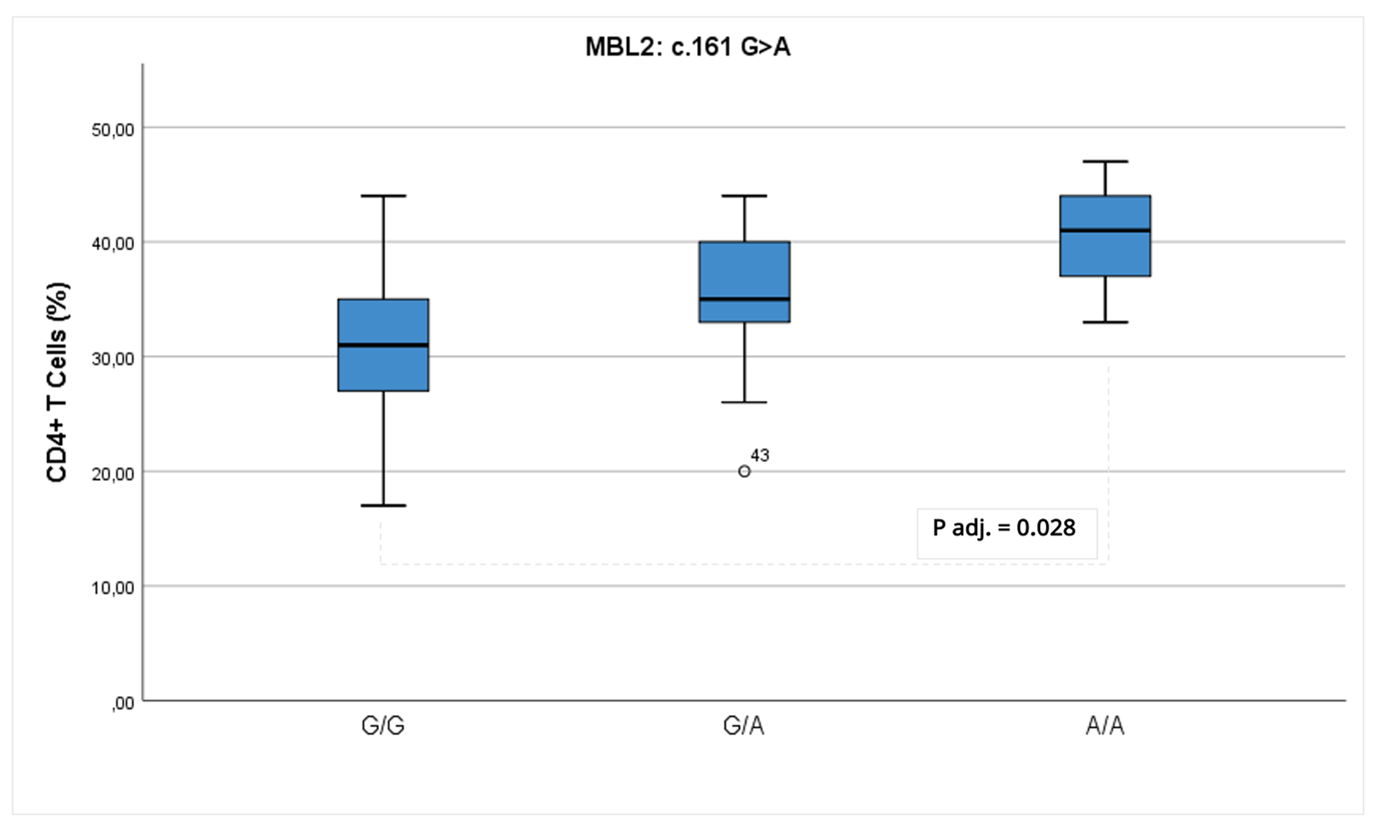
Figure 6. Immunophenotypic differences associated with MBL2 genotype. Comparison of CD4+ T cell percentage across MBL2 c.161 G>A genotypes.
After adjustment for age and sex, MBL2 c.161 G>A genotype remained significantly associated with CD4+ T-cell percentages (p=0.028). Estimated marginal means indicated higher CD4+ T-cell proportions in individuals carrying the A/A genotype compared to C/C carriers. In contrast, the previously observed association between MBL2 genotype and CD3−CD16+CD56+ NK cell counts were no longer statistically significant after adjustment, while age remained a significant predictor (p=0.01). Similarly, the lower frequency of effector CD8+ T cells observed in patients harboring MBL2 variants of uncertain significance did not remain significant after adjustment for age and sex. These findings suggest a selective association between MBL2 c.161G>A genotype and relative CD4+ T-cell distribution, whereas other observed differences appear to be driven predominantly by age-related effects (NK cells) or may reflect random variation rather than a robust genotype effect (effector CD8+ T cells).
Additionally, for 59 of the patients the genotype profile of a total of 17 targeted SNP variants in immune-related genes was achieved. The genotype and allele frequencies are presented in Supplementary Table 2; the reference allele was indicated as “N” and the alternate allele - as “M”, respectively. Additional information concerning the corresponding allele frequencies in the available databases is also provided. The allele frequency for the alternate allele of the selected variants in our cohort was compared with the allele frequency reported in the VARSOME database (http://varsome.com) for the Bulgarian controls and no statistically significant difference was achieved according to the Fisher’s exact test (Supplementary Table 2).
Discussion
Immunophenotyping in our cohort revealed significant abnormalities in the T, B, and NK cell populations, supporting the presence of underlying immunodeficiency in some children with recurrent infections. In particular, a reduced number of CD4+ T cells was observed. Given their central role in adaptive immunity, CD4+ T cell deficiency is associated with increased susceptibility to viral and opportunistic infections and is a common feature in combined or syndromic immunodeficiencies [8]. A reduction in total and memory B cells was also common. Impaired development of memory B cells is a hallmark of humoral immunodeficiencies such as common variable immunodeficiency (CVID), which is characterized by hypogammaglobulinemia, poor antibody responses, and recurrent respiratory infections [9]. These defects have also been found in monogenic forms of CVID, in which both B-cell and T-cell abnormalities contribute to immune dysregulation [10]. NK cell deficiencies, including reduced numbers and function, have been found in some patients. As key effectors against virus-infected and malignant cells, NK cell dysfunction has been associated with severe herpesvirus infections and impaired viral defense [11]. These findings are consistent with the existing literature and emphasize the role of NK cell dysregulation in recurrent infections associated with innate defects in immunity. Taken together, these abnormalities of lymphocyte subsets underscore the heterogeneity of immune dysfunction in affected children and support the diagnostic value of immunophenotyping in the assessment of suspected primary immunodeficiencies. This study intentionally included a clinically heterogeneous cohort representative of real-world pediatric referrals for recurrent infections and suspected immune dysfunction as the primary inclusion criterion. Children presenting with allergic, autoinflammatory, or autoimmune manifestations were included, as these phenotypes are increasingly recognized as part of the clinical spectrum of inborn errors of immunity and may precede or coexist with susceptibility to infection. This approach reflects current concepts of immune dysregulation and enhances the clinical relevance of the findings.
Age stratification was applied to reflect the substantial maturation of the immune system across pediatric developmental stages, from early childhood to adolescence. In addition, sex distribution was reported to account for potential sex-related differences in immune responses, particularly during puberty. Although the study was not powered to perform sex-specific subgroup analyses, inclusion of these demographic variables improves transparency and biological interpretability of the findings.
Immunoglobulin profiling showed a high frequency of decreased serum IgG and IgA in the patients. Such hypogammaglobulinemia is typical of primary antibody deficiencies such as common variable immunodeficiency (CVID) and selective IgA deficiency (sIgAD) [12]. CVID is associated with low IgG and either IgA or IgM deficiency and impairs response to vaccines, often leading to recurrent respiratory and gastrointestinal infections [13]. sIgAD, the most common primary immunodeficiency worldwide, is associated with mucosal infections, allergies and autoimmunity [14]. Some patients also showed significantly elevated IgE levels, suggesting hyper-IgE syndromes (HIES), which include autosomal dominant (STAT3 mutations) and autosomal recessive forms (DOCK8, PGM3-associated) [15]. HIES typically presents with eczema, recurrent skin and lung infections and IgE levels of >2,000 IU/ml. Immunoglobulin abnormalities are frequently found in children with recurrent respiratory infections, which underlines the diagnostic value of Ig serum screening [16]. Subclass deficiencies – particularly in IgG2 and IgA – are strongly associated with recurrent sinopulmonary infections in children. IgG2 is crucial for the opsonization of encapsulated bacteria such as Streptococcus pneumoniae and Haemophilus influenzae, while IgA protects the mucosal surfaces of the respiratory tract and gastrointestinal tract [17].
Repeated complete blood counts with differential, including absolute neutrophil counts (ANC), were available from hospital medical records and were obtained during both infectious episodes and clinically stable periods. Persistent neutropenia was not documented. Transient decreases in ANC were observed in a subset of patients, typically during acute infections or following antibiotic or viral exposure, with subsequent normalization on follow-up. This pattern is inconsistent with congenital or chronic neutropenia. Functional neutrophil testing (oxidative burst assays) was not routinely performed, as there was no laboratory or clinical phenotype suggestive of chronic granulomatous disease, including absence of invasive fungal infections, deep abscesses, or granulomatous inflammation. In addition, no clinical features characteristic of ELANE-associated or other congenital neutropenias were identified, and whole-exome sequencing did not reveal pathogenic variants in genes associated with inherited neutrophil disorders. Importantly, the cohort comprised children evaluated for recurrent infections rather than patients with established inborn errors of immunity. Neutrophil assessment therefore focused on baseline quantitative parameters, and the lack of persistent abnormalities did not warrant extended neutrophil-specific functional testing across the cohort.
Microbiological data from respiratory samples highlighted the predominance of bacterial pathogens, particularly Haemophilus influenzae and Staphylococcus aureus, which are frequently implicated in pediatric lower and upper respiratory tract infections. These bacteria are known to colonize mucosal surfaces and can lead to significant morbidity in children with underlying immune dysfunction. The identification of these pathogens reinforces their clinical relevance, especially in immunocompromised hosts where clearance is impaired. However, the high rate of negative culture results observed in our cohort may reflect several factors, including recent antibiotic exposure, non-culturable organisms, and infections of viral origin. The detection of vaccine-preventable organisms underscores the importance of adherence to national immunization schedules, particularly for Streptococcus pneumoniae and Haemophilus influenzae type b. Children with immunodeficiencies may not mount adequate vaccine responses, making herd immunity and booster strategies critical for protection. Revaccination protocols and the use of conjugate vaccines in at-risk populations should be prioritized to reduce the burden of preventable disease.
Our cohort comprised children referred for evaluation of recurrent infections, rather than those with a pre-established diagnosis of inborn errors of immunity. Consequently, immunological assessments were conducted during clinically stable periods, and patients were generally not acutely ill at the time of immune phenotyping. For this reason, routine respiratory viral panels (e.g. RSV, influenza, adenovirus) were not systematically performed during hospitalization, and their absence should be considered a limitation when interpreting immune activation patterns in this retrospective design. Molecular analysis for herpesvirus DNA was performed in a subset of patients: HHV-6 DNA was identified in three children, while HHV-7 DNA was detected in four children. EBV DNA was detected in five children; of these, four had low-level viral loads below the predefined quantitative threshold, and one had a higher viral load. CMV DNA and HSV-1/2 DNA were not detected. Overall, these data indicate that while respiratory viruses were not routinely assessed during immune-status admissions, selected herpesvirus PCR results were available and occasionally positive. We now report this transparently and will acknowledge it as a limitation regarding unmeasured acute respiratory viral exposures.
Based on expert review of hospital medical records, a history of SARS-CoV-2 infection was documented in a minority of patients: 11 patients (18.3%). When reported, COVID-19 was uniformly mild, managed on an outpatient basis, and not associated with hospitalization, oxygen requirement, or severe pulmonary involvement. In the remaining cases, no anamnestic data suggestive of clinically significant COVID-19 were recorded. SARS-CoV-2 PCR or antigen testing and COVID-19 vaccination status were not systematically documented, reflecting the retrospective design and the fact that hospital admissions were related to immunological evaluation rather than acute infection. Importantly, no cases of multisystem inflammatory syndrome in children (MIS-C) or long COVID were identified, and no persistent post-COVID symptoms or COVID-related immunological complications were documented.
Biochemical analyses revealed a trend toward decreased serum levels of 25(OH)-vitamin D. Vitamin D plays a regulatory role in innate and adaptive immunity, including the modulation of antimicrobial peptide expression and cytokine production. Its deficiency has been linked to increased susceptibility to infections, immune dysregulation, and poor vaccine responses [18]. Hypocomplementemia, especially low C4, may signal underlying complement pathway defects or consumption due to immune complex formation, and is often associated with autoimmune conditions such as systemic lupus erythematosus. While most patients had normal C3 levels and negative ANA tests, these findings warrant further evaluation using extended autoantibody panels, and clinical correlation to rule out evolving autoimmune phenomena.
Genetic analysis through whole-exome sequencing revealed clinically significant variants in several genes linked to primary immunodeficiencies and immune dysregulation, particularly TNFRSF13B, IL10RA, and CFTR. Mutations in TNFRSF13B, which encodes the transmembrane activator and calcium modulator and cyclophilin ligand interactor, have been well-documented in association with CVID, especially in patients presenting with hypogammaglobulinemia, impaired memory B cell formation, and recurrent sinopulmonary infections [19]. While the pathogenicity of TNFRSF13B variants can vary based on zygosity and coexisting mutations, their presence supports a diagnosis of humoral immunodeficiency. Variants in IL10RA, encoding the alpha subunit of the interleukin-10 receptor, have been implicated in early-onset inflammatory bowel disease and systemic inflammation. Their identification in our cohort may reflect underlying immune dysregulation, consistent with recent findings of IL10RA mutations contributing to unrestrained inflammatory responses due to defective regulatory cytokine signaling [20]. Additionally, CFTR variants – commonly associated with cystic fibrosis – were observed in some patients. It was speculated that even in heterozygous states, CFTR mutations can predispose to recurrent airway infections and altered mucosal immunity, suggesting a broader immunomodulatory role than previously recognized [21].
Interestingly, although MBL2 variants detected in our case-group were classified as variants of uncertain significance, immunophenotypic analysis revealed measurable differences in immune cell distribution. Children with the A/A genotype of the MBL2 c.161 G>A variant exhibited elevated CD4+ T cell percentages independent of age and sex but lower NK cell counts, suggesting that MBL2 polymorphisms may influence the balance between adaptive and innate immune responses [22]. NK-cell levels are strongly age-dependent, which attenuated the apparent association with MBL2 genotype after adjustment. While further functional validation is needed, these findings highlight the potential of MBL2 polymorphisms to affect immune homeostasis [23].
Beyond monogenic causes, targeted SNP analysis revealed genetic profile of some variants in genes critical for innate immunity, such as TLR4, TLR9, etc. These genes encode pattern recognition receptors and inflammatory mediators that are essential for the early immune response to pathogens. Polymorphisms in TLR4 and TLR9 have been reported to be associated with altered responses to bacterial and viral components, leading to either hyperinflammatory reactions or blunted immune defenses [24]. Variants in VEGFA and MAP3K1 were also analyzed, pointing toward disturbances in tissue repair and epithelial barrier integrity. VEGFA encodes vascular endothelial growth factor A, a key regulator of angiogenesis and mucosal healing, while MAP3K1 is involved in intracellular signaling cascades related to cell survival and inflammation. Alterations in these genes have been linked to increased susceptibility to infections and defective resolution of inflammatory responses, particularly in the respiratory tract [25].
Genetic analysis revealed some genetic variants associated with a broad spectrum of monogenic and polygenic conditions in the study cohort, highlighting the possible heterogeneity of underlying etiologies in children with recurrent infections. Among the diagnosed disorders there were several with known immunologic, autoinflammatory, dermatological, and hematological implications. A subset of patients was diagnosed with autosomal recessive congenital ichthyosis type 3 (OMIM: 606545) and ichthyosis vulgaris (OMIM: 146700), in both autosomal dominant and recessive forms. These disorders affect skin barrier function and may predispose individuals to secondary infections due to epithelial compromise. Similarly, Netherton syndrome (OMIM: 256500), a severe autosomal recessive genodermatosis, was identified and is known to be associated with recurrent infections and elevated IgE levels due to a defective skin barrier and immune dysregulation. Neurological syndromes were also identified, including autosomal dominant nocturnal frontal lobe epilepsy type 4 (OMIM: 610353), which, although primarily a neurologic condition, can occasionally coexist with immune phenotypes, particularly in syndromic presentations. Autoinflammatory and immunoregulatory disorders were represented by findings such as familial periodic fever with autosomal dominant inheritance (OMIM: 142680), and pyogenic sterile arthritis, pyoderma gangrenosum, and acne (PAPA syndrome; OMIM: 604416). Both syndromes are characterized by recurrent inflammation and can mimic or overlap with primary immunodeficiencies in clinical presentation. Several hematologic and iron-related disorders were also identified: beta-thalassemia (OMIM: *141900 and *141900+ -plus), congenital dyserythropoietic anemia type IIIA (OMIM: 105600), and hemochromatosis type 1 (OMIM: 235200). These may present with anemia or iron overload and may complicate infection susceptibility due to altered host-pathogen interactions.
Importantly, several children were found to have variants in immunodeficiency-related genes:
- STAT3-related disease (OMIM: *102582), known to include autosomal dominant hyper-IgE syndrome and immunodeficiency phenotypes.
- Common variable immunodeficiency (CVID) types 2 (OMIM: 240500), AD/AR (gene: TNFRSF13B) and 13 (OMIM: 616873), AD (gene: IKZF1) – with autosomal dominant inheritance.
- Fanconi anemia group I (OMIM: 609053), AR (gene: FANCI) associated with chromosomal instability and increased susceptibility to infection and malignancy.
- Centromeric instability–facial anomalies–immunodeficiency syndrome (OMIM: 242860), AR (gene: DNMT3B) – a rare recessive condition affecting immune and developmental pathways.
- IL10RA and TNFRSF13B variants were also observed in other parts of the cohort, supporting their diagnostic relevance.
Some genetic variants detected in STAT3-related disease, TNFRS13B, IL10RA, etc. may present with autoimmunity and/or inflammation rather than infections. Therefore, it will be beneficial to perform a comparison study to identify the possible different prevalence of targeted variants between children with and without recurrent infections.
Some patients were diagnosed with X-linked conditions, including hemophilia A (OMIM: 306700) and thrombophilia 13 (OMIM: 301071), associated gene: F8 – potentially complicating infections with bleeding diatheses or thromboses.
Other non-immunologic conditions of relevance included:
- Renal glucosuria (OMIM: 233100), AD/AR (gene: SLC5A2) – in both dominant and recessive forms.
- Wilson’s disease (OMIM: 277900), AR (gene: ATP7B) – a copper metabolism disorder.
- GJB3-related hearing loss (OMIM: 603324), AD/AR, (gene: GJB3).
- Cancer susceptibility syndromes, including familial predisposition to breast and ovarian cancer (OMIM: 604370), AD and pancreatic cancer type 4 (OMIM: 614320) – (gene: BRCA1).
Notably, most of these diagnoses were only possible through whole-exome sequencing, demonstrating the value of genomic tools in unraveling the complex landscape of recurrent infections in children.
For the remaining children with variants of uncertain significance, close clinical and immunological monitoring will be maintained. As variant reclassification evolves through international databases and novel genotype-phenotype correlations, future diagnostic updates will be performed to refine patient management and prognosis.
These results underline the importance of early immunological evaluation in children with recurrent infections. However, this study has several limitations. The cohort was relatively small and from a single clinical center, which may limit the applicability of the results to wider pediatric populations. Functional validation of many variants of uncertain significance (VUS) was not possible, and segregation analysis was incomplete in some cases due to missing parental samples. Environmental and nutritional factors were not systematically analyzed, and prior use of antibiotics may have interfered with pathogen detection. These limitations underline the need for multicenter studies, longer follow-up and functional testing to improve genotype-phenotype interpretation.
Conclusion
The integration of immunophenotyping, immunoglobulin profiling, single nucleotide polymorphism analysis, and whole-exome sequencing in this pediatric cohort with recurrent infections has provided valuable insights into the diverse immunological and genetic mechanisms underlying susceptibility to infection. This multifaceted approach enabled the stratification of patients into distinct immunological subgroups, each with potential therapeutic and prognostic implications. One subset of patients displayed features consistent with classical primary immunodeficiency, including profound adaptive immune deficits requiring interventions such as immunoglobulin replacement therapy and antimicrobial prophylaxis. Another subgroup exhibited innate immune dysfunction, marked by SNPs in genes involved in pathogen recognition and inflammatory signaling. These children may benefit from immunomodulatory therapies targeting specific innate immune pathways. Additionally, a portion of the cohort demonstrated clinical and genetic features suggestive of autoinflammatory syndromes – particularly involving MEFV or NLRP variants – potentially warranting targeted biologic therapies such as IL-1 inhibitors. A final subgroup appeared to suffer from secondary immune impairment, likely influenced by environmental, nutritional, or metabolic factors, underscoring the importance of supportive care strategies including vaccination, nutritional supplementation, and environmental risk mitigation. These findings highlight the necessity of a personalized, systems-based diagnostic framework that combines immunologic evaluation with genomic data. Functional assays, including vaccine response testing and cytokine profiling, should be incorporated into diagnostic workflows to refine assessments of immune competence. Variants of uncertain significance identified through genetic testing merit continued investigation through longitudinal follow-up and functional validation studies.
In conclusion, this study underscores the clinical utility of combining immunological and genomic data to guide precision diagnostics and individualized treatment in children with recurrent infections. Such an approach not only facilitates earlier and more accurate diagnosis but also enables the development of tailored therapeutic strategies, ultimately improving outcomes and quality of life in this vulnerable population.
Conflict of Interest
The authors have no conflicts of interest to declare.
Informed Consent Statement
Informed consent was obtained from all parents of the patients involved in the study. None of the patients can be identified in this publication.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
Author Contributions
Conceptualization: P.K.; methodology: S.A., A.T., T.T., S.A.,M.S., Z.P., S.K., S.L., N.G.; software: P.K., T.L., S.A., A.T., T.T., S.A.,M.S., Z.P.; validation: P.K., T.L., and G.P.; formal analysis: S.A., T.L.; investigation: P.K., T.L., S.A., G.P.; resources: P.K., T.L., S.A., G.P.; data curation: P.K., T.L., S.A., G.P; writing – original draft preparation: P.K.; writing – review and editing: P.K., T.L., G.P., S.A; visualization: P.K., T.L.; supervision: P.K., G.P.; project administration: P.K.; funding acquisition: P.K., G.P. All authors have read and agreed to the published version of the manuscript.
Abbreviations
CVID: Common Variable Immunodeficiency; IEIs: Inborn Errors of immunity; PIDs: Primary Immunodeficiency Disorders; SCID: Severe Combined Immunodeficiency; SNP: Single Nucleotide Polymorphism; WES: Whole Exome Sequencing; XLA X: linked agammaglobulinemia.
List of Genes Included in Gene Panel “Immune Deficiency”
ACD, ACP5, ACTB, ADA, ADAM17, ADAR, AICDA, AIRE, AK2, ALPI, AP3B1, AP3D1, ARHGEF1, ARPC1B, ATM, ATP6AP1, B2M, BACH2, BCL10, BCL11B, BLM, BLNK, BTK, C17ORF62, C1QA, C1QB, C1QC, C1S, C2, C3, C5, C6, C7, C8A, C8B, C9, CARD11, CARD14, CARD9, CASP10, CASP8, CCBE1, CD19, CD247, CD27, CD3D, CD3E, CD3G, CD4, CD40, CD40LG, CD46, CD55, CD59, CD70, CD79A, CD79B, CD81, CD8A, CDC42, CDCA7, CDK9, CEBPE, CECR1, CFB, CFD, CFH, CFI, CFP, CFTR, CHD7, CIITA, CLCN7, CLPB, COG6, COLEC11, COPA, CORO1A, CR2, CSF2RA, CSF2RB, CSF3R, CTC1, CTLA4, CTPS1, CTSC, CXCR4, CYBA, CYBB, CYP27A1, DBR1, DCLRE1C, DDX58, DGAT1, DGKE, DIAPH1, DKC1, DNAJC21, DNASE1L3, DNASE2, DNMT3B, DOCK2, DOCK8, DSG1, EFL1, ELANE, EPG5, ERCC6L2, EXTL3, FADD, FANCA, FAS, FASLG, FAT4, FCGR3A, FCHO1, FERMT3, FOXN1, FOXP3, G6PC, G6PC3, G6PD, GATA2, GFI1, GINS1, GUCY2C, HAVCR2, HAX1, HELLS, HMOX1, HYOU1, ICOS, IFIH1, IFNAR2, IFNGR1, IFNGR2, IGLL1, IKBKB, IKZF1, IL10, IL10RA, IL10RB, IL12B, IL12RB1, IL17RA, IL17RC, IL1RN, IL21, IL21R, IL23R, IL2RA, IL2RB, IL2RG, IL36RN, IL6R, IL6ST, IL7, IL7R, IRAK4, IRF2BP2, IRF4, IRF7, IRF8, ISG15, ITGB2, ITK, JAGN1, JAK1, JAK3, KRAS, LAMTOR2, LAT, LCK, LIG1, LIG4, LPIN2, LRBA, LYST, MAGT1, MALT1, MAP3K14, MASP1, MEFV, MKL1, MOGS, MRE11A, MSN, MTHFD1, MVK, MYD88, MYO5A, NBN, NCF1, NCF2, NCF4, NCSTN, NFE2L2, NFKB1, NFKB2, NFKBIA, NHEJ1, NHP2, NLRC4, NLRP1, NLRP12, NLRP3, NOD2, NOP10, NRAS, NSMCE3, OBFC1, OFD1, ORAI1, OTULIN, PARN, PEPD, PGM3, PIGA, PIK3CD, PIK3R1, PLCG2, PMS2, PNP, POLA1, POLD1, POLE, POLE2, POMP, PRF1, PRG4, PRKCD, PRKDC, PSENEN, PSMB4, PSMB8, PSTPIP1, PTPRC, RAB27A, RAC2, RAG1, RAG2, RANBP2, RASGRP1, RBCK1, RECQL4, RELA, RELB, RFX5, RFXANK, RFXAP, RHOH, RIPK1, RLTPR, RMRP, RNASEH2A, RNASEH2B, RNASEH2C, RNF168, RNF31, RNU4ATAC, RORC, RPSA, RTEL1, SAMD9, SAMD9L, SAMHD1, SBDS, SEC61A1, SERPING1, SH2D1A, SLC29A3, SLC35C1, SLC37A4, SLC39A7, SLC46A1, SLC7A7, SMARCAL1, SMARCD2, SP110, SPINK5, SPPL2A, SRP54, SRP72, STAT1, STAT2, STAT3, STAT5B, STIM1, STK4, STX11, STXBP2, TAP1, TAP2, TAPBP, TAZ, TBX1, TCF3, TCN2, TERC, TERT, TFRC, TGFB1, THBD, TINF2, TLR3, TMC6, TMC8, TMEM173, TNFAIP3, TNFRSF13B, TNFRSF1A, TNFRSF4, TNFRSF9, TRAF3IP2, TREX1, TRNT1, TTC37, TTC7A, TYK2, UBA1, UNC119, UNC13D, UNC93B1, UNG, USB1, USP18, VPS13B, VPS45, WAS, WDR1, WIPF1, WRAP53, XIAP, ZAP70, ZBTB24, ZNF341.
Acknowledgements
The authors thank the patients and their families for participating in this study. This work was supported by Medical University Sofia.
Funding Statement
This research was partly funded by:
- A grant from the Medical University of Sofia (Council of Medical Science, project no. 7343/2021, grant no. 170/2022).
- A grant from the Medical University of Sofia (Council of Medical Science, grant no. D156/29.05.2024).
ORCID
P Kostova: http://orcid.org/0000-0002-3012-1861.
References
2. Tangye SG, Al-Herz W, Bousfiha A, Cunningham-Rundles C, Franco JL, Holland SM, et al, Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022 Oct;42(7):1473–507.
3. Stiehm ER, Ochs HD, Winkelstein JA, editors. Stiehm’s Immune Deficiencies. 2nd ed. London: Academic Press; 2014.
4. Oliveira JB, Fleisher TA. Laboratory evaluation of primary immunodeficiencies. J Allergy Clin Immunol. 2010 Feb;125(2 Suppl 2):S297–305.
5. Pieniawska-Śmiech K, Pasternak G, Lewandowicz-Uszyńska A, Jutel M. Diagnostic Challenges in Patients with Inborn Errors of Immunity with Different Manifestations of Immune Dysregulation. J Clin Med. 2022 Jul 20;11(14):4220.
6. Chinn IK, Chan AY, Chen K, Chou J, Dorsey MJ, Hajjar J, et al. Diagnostic interpretation of genetic studies in patients with primary immunodeficiency diseases: A working group report of the Primary Immunodeficiency Diseases Committee of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2020 Jan;145(1):46–69.
7. Dyer SC, Austine-Orimoloye O, Azov AG, Barba M, Barnes I, Barrera-Enriquez VP, et al. Ensembl 2025. Nucleic Acids Res. 2025 Jan 6;53(D1):D948–57.
8. Kalina T, Bakardjieva M, Blom M, Perez-Andres M, Barendregt B, Kanderová V, et al. EuroFlow Standardized Approach to Diagnostic Immunopheneotyping of Severe PID in Newborns and Young Children. Front Immunol. 2020 Mar 19;11:371.
9. Ahn S, Cunningham-Rundles C. Role of B cells in common variable immune deficiency. Expert Rev Clin Immunol. 2009 Sep;5(5):557–64.
10. Azizi G, Hesari MF, Sharifinejad N, Fayyaz F, Chavoshzadeh Z, Mahdaviani SA, et al. The Autoimmune Manifestations in Patients with Genetic Defects in the B Cell Development and Differentiation Stages. J Clin Immunol. 2023 May;43(4):819–34.
11. Della Chiesa M, De Maria A, Muccio L, Bozzano F, Sivori S, Moretta L. Human NK Cells and Herpesviruses: Mechanisms of Recognition, Response and Adaptation. Front Microbiol. 2019 Oct 4;10:2297.
12. Hammarström L, Vorechovsky I, Webster D. Selective IgA deficiency (SIgAD) and common variable immunodeficiency (CVID). Clin Exp Immunol. 2000 May;120(2):225–31
13. Cunningham-Rundles C. Common variable immune deficiency: case studies. Blood. 2019 Nov 21;134(21):1787–95.
14. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand J Immunol. 2017 Jan;85(1):3–12.
15. Kasap N, Celik V, Isik S, Cennetoglu P, Kiykim A, Eltan SB, et al. A set of clinical and laboratory markers differentiates hyper-IgE syndrome from severe atopic dermatitis. Clin Immunol. 2021 Feb;223:108645.
16. Peddi NC, Vuppalapati S, Sreenivasulu H, Muppalla SK, Reddy Pulliahgaru A. Guardians of Immunity: Advances in Primary Immunodeficiency Disorders and Management. Cureus. 2023 Sep 7;15(9):e44865.
17. Abolnezhadian F, Saki N, Nikakhlagh S, Safavi E, Asar S. The relative frequency of primary immunodeficiency diseases in pediatric patients with recurrent sinusitis and otitis media. Electron J Gen Med. 2019 Jan 1;16(1):em102.
18. Ismailova A, White JH. Vitamin D, infections and immunity. Rev. Endocr. Metab. Disord. 2022 Apr;23(2):265–77.
19. Sathkumara HD, De Silva NR, Handunnetti S, De Silva AD. Genetics of common variable immunodeficiency: role of transmembrane activator and calcium modulator and cyclophilin ligand interactor. Int. J. Immunogenet. 2015 Aug;42(4):239–53.
20. Aschenbrenner D, Ye Z, Zhou Y, Hu W, Brooks I, Williams I, et al. Pathogenic Interleukin-10 Receptor Alpha Variants in Humans - Balancing Natural Selection and Clinical Implications. J Clin Immunol. 2023 Feb;43(2):495–511.
21. Zhang X, Moore CM, Harmacek LD, Domenico J, Rangaraj VR, Ideozu JE, et al. CFTR-mediated monocyte/macrophage dysfunction revealed by cystic fibrosis proband-parent comparisons. JCI Insight. 2022 Mar 22;7(6):e152186.
22. Queiroz MAF, Santiago AM, Brito WRDS, Pereira KAS, de Brito WB, Torres MKDS, et al. Polymorphisms in the MBL2 gene are associated with the plasma levels of MBL and the cytokines IL-6 and TNF-α in severe COVID-19. Front Immunol. 2023 Apr 17;14:1151058.
23. Kalia N, Singh J, Kaur M. The ambiguous role of mannose-binding lectin (MBL) in human immunity. Open Med (Wars). 2021 Feb 17;16(1):299–310.
24. Medvedev AE. Toll-like receptor polymorphisms, inflammatory and infectious diseases, allergies, and cancer. J Interferon Cytokine Res. 2013 Sep;33(9):467–84.
25. Cheng Y, Sun Y, Ji Y, Jiang D, Teng G, Zhou X, et al. Novel compound variants of the AR and MAP3K1 genes are related to the clinical heterogeneity of androgen insensitivity syndrome. Biosci Rep. 2020 May 29;40(5):BSR20200616.