Abstract
Telomeres are repetitive DNA sequences located at chromosomal ends that are crucial for maintaining genomic stability. Telomere lengths are tightly regulated under physiological conditions, disruption of which results in telomere shortening that ultimately leads to telomere biology disorders, such as Dyskeratosis congenita (DC), bone marrow failure syndromes, and Idiopathic Pulmonary Fibrosis (IPF), amongst others. Progressive telomere shortening is also a well-recognized feature of aging. Although telomere shortening and anemia often go hand-in-hand in aging individuals and Myelodysplastic Syndrome (MDS) patients, the mechanistic link in between these manifestations remains poorly understood. In this regard, RIOK2 has recently emerged as a key transcriptional regulator of both blood cell development and telomerase activity. This commentary elaborates the multifaceted roles of RIOK2 in maintaining hematopoiesis and telomere length homeostasis in the context of aging, MDS and IPF, and further explores the potential of regulating RIOK2’s expression and/or function for therapeutic applications.
Keywords
RIOK2, Telomere, MDS, Aging, Telomerase, IPF
Review
Telomeres, located at chromosomal ends, are repetitive genomic DNA sequences surrounded by proteins that shield them from being recognized as DNA double strand breaks, thereby preventing a resultant DNA damage response [1-3]. In a vast majority of proliferative cells, telomeric DNA is replicated by telomerase, a ribonucleoprotein complex responsible for de novo generation of telomeric DNA via reverse transcription [4]. Telomerase is a highly complex structure consisting of the telomerase reverse transcriptase (TERT), an RNA template (TERC), and several protein cofactors that are involved in the assembly, stability, trafficking and proper functioning of the telomerase [5]. Telomere length is tightly regulated by the balance between telomere elongation and shortening mechanisms [6-9], and disruption of either has distinct pathological consequences [9,10]. In physiological aging, telomeres progressively shorten, until they become too short, thus triggering senescence or apoptotic pathways. As a consequence, telomere shortening is a hallmark of aging and is implicated in the etiology of multiple age-related diseases [1,11]. Shortening of telomeres is also observed in a spectrum of diseases collectively known as telomere biology disorders (TBDs), affecting multiple organ systems with a wide scale of clinical presentations [12]. TBDs are often associated with increased morbidity, and their phenotypes range from idiopathic pulmonary fibrosis (IPF) to bone marrow failure and cryptogenic liver cirrhosis [13].
Although breakthrough discoveries regarding the structure, function, and regulation of these highly complex structures have been made over the past decades, significant gaps remain in our understanding of the regulation of telomere homeostasis, the underlying mechanisms of telomere dysfunction, patterns of organ impairment and the full spectrum of associated diseases. Addressing these knowledge gaps is crucial as TBDs are associated with premature mortality, lack effective biomarkers and treatments, and currently rely on transplantation as the only cure [14]. Organ transplantation carries significant morbidity and mortality even in non-TBD patients, with the risk being markedly higher in the TBD patient group. For example, a shorter median survival is reported in TBD lung transplant recipients compared to non-TBD patients. Several post-transplant complications occurred at a higher frequency after lung transplantation in IPF patients harboring telomerase mutations [14,15] and uncommon complications were observed after bone marrow transplantation in Dyskeratosis congenita (DC), including late vascular complications and Evans syndrome [16].
Bone marrow failure is a common presentation of TBDs, and myelodysplastic syndromes (MDS) are often associated with bone marrow failure. Ineffective hematopoiesis and morphological abnormalities of myeloid cell lines are the major factors in MDS pathogenesis. Clinically, it is characterized by peripheral blood cytopenia and an increased risk of transformation into acute myeloid leukemia (AML) [17-19]. While this transformation is an important cause of mortality, other major causes of death in this patient group are the complications of MDS itself, such as infections [20]. Risk of MDS increases with age, with a median age of approximately 70 at the time of diagnosis [17,21]. The disease encompasses a spectrum of phenotypes, marked by significant heterogeneity in clinical course and long-term outcomes [22]. Consequently, risk stratification using prognostic tools (e.g. International Prognostic Scoring System [IPSS], Revised IPSS [IPSS-R], molecular IPSS [IPSS-M]) is critical for guiding management [23]. For lower-risk MDS (LR-MDS; low/intermediate-1), as per IPSS criteria, priorities include improving cytopenias and reducing transfusion dependency to enhance quality of life. In contrast, treatment for higher-risk MDS (HR-MDS) focuses on delaying disease progression and improving survival [24]. Although various treatment approaches aim to achieve these goals, hematopoietic stem cell transplantation (HSCT) remains the only curative option. Efforts to identify alternative therapeutic targets in the pathogenesis of MDS are actively ongoing to improve outcomes.
Recent advancements in understanding MDS pathogenesis revealed an association between this disease and telomere homeostasis. In several studies, a remarkable proportion of MDS patients have been found to have significantly shorter telomeres compared to controls, and it has been reported that telomere length has critical implications for prognosis and treatment outcome [25-28]. Myllymäki et al. reported that telomere length in recipient blood is a significant predictor of survival and non-relapse mortality after hematopoietic stem cell transplantation in MDS patients older than 40 years of age [27]. Göhring et al. suggested that shorter telomeres might be predictive of increased risk of leukemic transformation [29]. Interestingly, there is a high burden of short telomeres in MDS patients, but relatively few mutations have been identified in the telomerase complex genes, thus strongly suggesting the presence of additional mechanisms behind telomere shortening in MDS [30,31].
The telomerase complex has thus become an attractive therapeutic target for MDS and other diseases associated with dysregulated telomere homeostasis. The therapeutic approach varies, aiming to enhance telomerase activity in conditions such as dyskeratosis congenita [32], and inhibit it in others, including many cancers [33]. A first-in-class therapy, imetelstat is an oligonucleotide telomerase inhibitor [34,35] that is recently approved by the Food and Drug Administration (FDA) for adults with LR-MDS who have transfusion-dependent anemia [36]. Specifically, this includes patients who require four or more red blood cell units over eight weeks and who have relapsed, are refractory, or are ineligible for erythropoiesis-stimulating agents (ESAs) [36]. The Phase 3 IMerge study results demonstrated imetelstat's efficacy in achieving clinically significant transfusion independence and improving hematologic response in this patient group [37]. These patients were naïve to lenalidomide, hypomethylating agents and did not harbor the del(5q) chromosomal abnormality. According to the study results, cytopenias—primarily neutropenia and thrombocytopenia, were the major adverse effects of imetelstat [37].
Using a telomerase inhibitor in the setting of shortened telomeres might seem paradoxical. However, despite shortened telomeres, telomerase activity in MDS is reported to be highly heterogenous [38]. A study by Ohyashiki et al. showed normal-to-low levels of telomerase activity in the bone marrow and peripheral blood mononuclear cells of most MDS patients, except for one patient with refractory anemia (RA) and two patients with refractory anemia with excess blasts in transformation (RAEBt) according to the French-American-British (FAB) classification [39]. These three patients, who exhibited high telomerase activity, subsequently progressed to acute leukemia. Conversely, several studies have shown significantly increased telomerase activity in the peripheral blood mononuclear cells of MDS patients compared to healthy controls [40]. Imetelstat selectively targets malignant progenitor cell clones with elevated telomerase activity compared to healthy counterparts, thus inducing apoptosis in these clones [37].
The heterogeneity of MDS, driven by diverse cytogenetic abnormalities, subtypes, and risk groups, underscores the need for individualized assessment. This is particularly relevant when telomerase-targeting approaches are considered given the variability in the reports of telomerase activity in MDS. Indeed, the analysis of the initial cohort of phase 2 studies on imetelstat found higher response rates in patients without the del(5q) chromosomal abnormality and who were naïve to lenalidomide and hypomethylating agents, prompting the expansion of cohorts focused on this patient group [41]. These findings highlight the critical role of abnormal telomere homeostasis in MDS pathogenesis and underscore the need for further exploration of telomere biology to identify novel therapeutic targets and biomarkers.
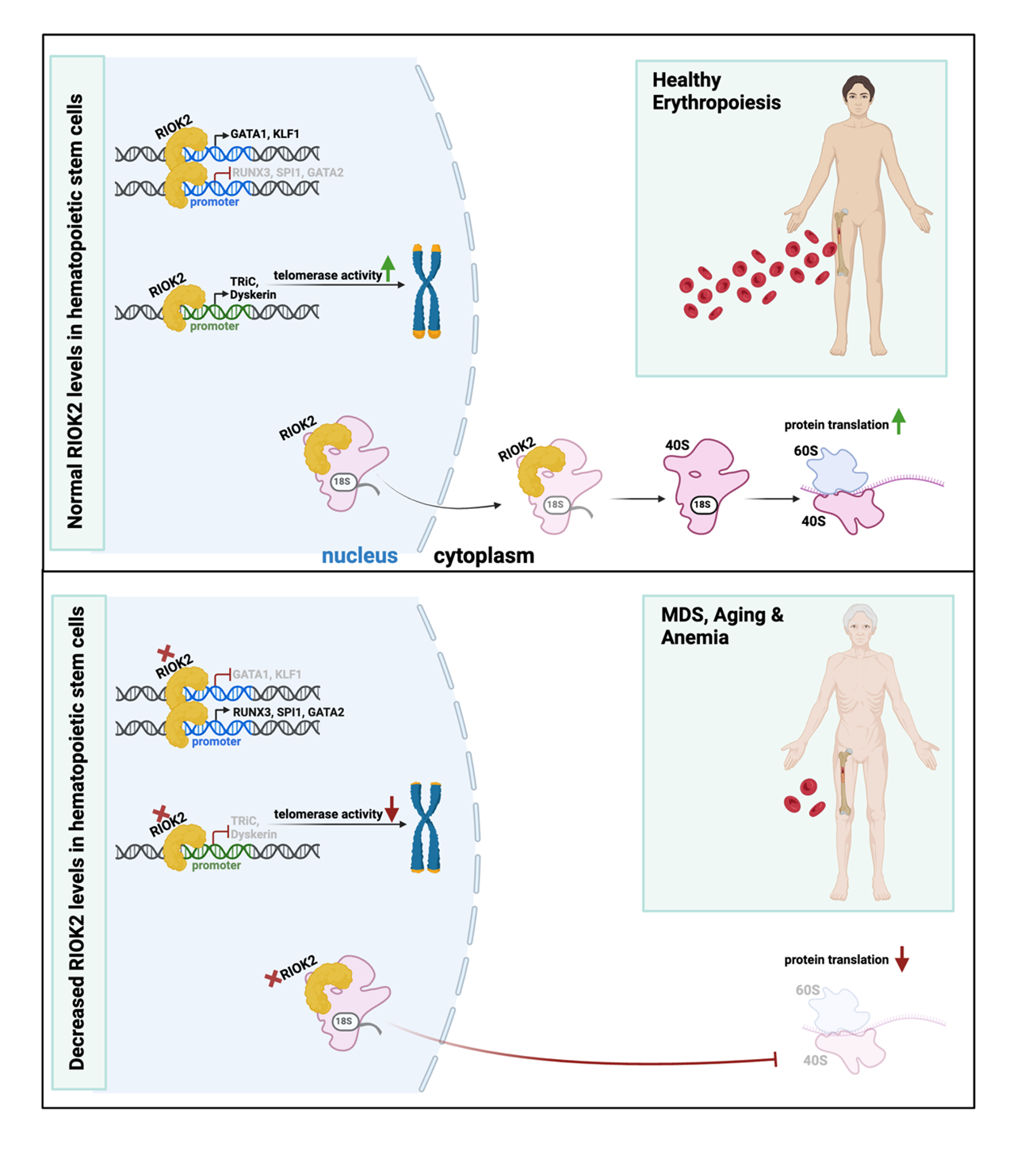
RIOK2 (right open reading frame kinase 2) is an atypical protein kinase deemed critical for ribosomal biogenesis and cytoplasmic protein translation [42]. It exhibits ubiquitous cytoplasmic expression across various human tissues [43], reflecting its fundamental role in cellular processes. To date, there are no reports of sex variations in RIOK2 expression patterns or sex-specific functional differences in humans. Sex-biased differences in disease settings are not reported so far either [44]. Recently, RIOK2 has been discovered to be a master transcription factor for hematopoietic lineage commitment. It has been shown that loss of RIOK2 dampens erythropoiesis while increasing megakaryopoiesis and myelopoiesis in primary human hematopoietic stem and progenitor cells (HSPCs). Particularly, the newly discovered N-terminal winged helix-turn-helix (wHTH) domain of RIOK2 enables it to bind DNA and act as a transcription factor critical for hematopoietic lineage commitment. Through this binding, RIOK2 fine-tunes lineage commitment by differentially regulating the expression of the transcription factors GATA1, GATA2, SPI1, RUNX3, and KLF1. It activates the mRNA expression of erythropoiesis-related genes, such as GATA1 and KLF1, while downregulating SPI1, RUNX3, and GATA2, that are genes associated with megakaryopoiesis or myelopoiesis. Thus, RIOK2 promotes erythropoiesis at the expense of myelopoiesis and megakaryopoiesis [45]. This partly explains the phenotypes of anemia and myeloproliferation in mice that harbor heterozygous loss of Riok2 in their hematopoietic compartment [45,46]. In this regard, RIOK2 is a rare protein with direct roles in transcription as well as translation. An important clue to its potential association with MDS, beyond the symptomatology resulting from its loss, is its location at human chromosome 5q15, a region adjacent to the commonly deleted region (CDR) in del(5q) subtype of MDS [46]. Indeed, RIOK2 mRNA levels correlate positively with erythroid gene expression and negatively with myeloid and megakaryocytic gene expression in MDS patient-derived bone marrow cells [45].
Interestingly, Ghosh et al. recently identified RIOK2 to be an upstream regulator of telomerase complex components TRiC and dyskerin [47]. TRiC is a chaperonin required for folding the telomerase cofactor TCAB1, which controls telomerase trafficking to Cajal bodies [48,49]. Mutations in TCAB1 interfere with its folding by TRiC and that leads to mislocalization of telomerase [48,50]. According to Freund et al., depletion of any one of the eight subunits of TRiC is sufficient to strongly decrease TCAB1 protein levels [48]. Loss of TRiC complex subunits inhibits TCAB1 biogenesis, recruitment of telomerase to telomeres and interferes with telomere lengthening, resulting in shortened telomeres [48]. In addition to its effects on telomerase trafficking, complete loss of TCAB1 results in decreased telomerase activity, thus demonstrating TCAB1’s critical functions in maintaining catalytic activity of the telomerase enzyme [51]. On the other hand, dyskerin is critical for the stabilization and maintenance of TERC [52,53]. Its loss results in defective telomerase activity and telomere shortening [54,55]. Upstream regulation of these components is largely undiscovered in contrast to TERT and TERC, two major core components of the telomerase complex that are thoroughly studied [56,57].
Ghosh et al. showed that loss of RIOK2 significantly reduces cellular proliferation and downregulates key genes involved in telomere maintenance in various erythroblast cell lines and primary human HSPCs, leading to telomere shortening. Additionally, RIOK2’s mRNA expression positively correlated with telomere length in MDS patient-derived bone marrow cells, associating shortened telomeres with reduced RIOK2 levels in these patients. Specifically, mRNA levels of TRiC and dyskerin complex subunits were shown to be downregulated upon loss of RIOK2 in HSPCs, in contrast to shelterin and CTC complex subunits that remained unaffected. Chromatin immunoprecipitation and luciferase reporter assays confirmed that RIOK2 transcriptionally regulates the expression of TRiC and dyskerin complex subunits. Mechanistically, RIOK2 binds to the promoter regions of CCT1 and CCT8, subunits of the TRiC complex, and induces their mRNA expression. This, in turn, enhances TCAB1 protein stability, as the TRiC complex facilitates proper folding of TCAB1 [48]. Additionally, RIOK2 binds to the promoters of dyskerin complex subunits, DKC1 and NHP2, and transactivates their mRNA expression. Consequently, loss of RIOK2 dampens expression of DKC1 and NHP2, that ultimately destabilizes TERC. The authors showed that loss of RIOK2 causes marked impairment of telomerase activity, as a result of diminished TCAB1 and TERC stability. In contrast, loss of RIOK2 did not perturb the mRNA expression of shelterin and CTC complex subunits. Thus, RIOK2 transcriptionally regulates TRiC and dyskerin complex subunits to promote telomerase activity and maintain telomere homeostasis. Of note, the authors have demonstrated that older individuals express lower levels of RIOK2 in their peripheral blood cells, in positive correlation with lower levels of TRiC and dyskerin complex subunits, as compared to younger individuals. This substantiates RIOK2’s relevance in telomere maintenance in physiological aging. Similar associations were also observed in IPF patient datasets, another age-associated telomere biology disorder. In addition, a positive correlation was reported between RIOK2 transcript levels and prognostic factors of survival in this patient cohort. At the cellular level, IPF patient-derived fibroblasts showed reduced mRNA expression of RIOK2 as compared to their control counterparts. Although there are different reports suggesting heterogeneity of IPF lung fibroblasts, several studies demonstrated that IPF lung fibroblasts have reduced proliferation rates compared to healthy lung fibroblasts [58-60]. Therefore, these data suggest similar negative effects of RIOK2’s loss on cell proliferation in IPF fibroblasts and erythroblast cell lines; this, however, needs to be confirmed in the same experimental setting. Nevertheless, Ghosh et al. showed that exogenous expression of RIOK2 markedly rescued telomere shortening in IPF patient-derived primary lung fibroblasts as compared to control lung fibroblasts [47]. It would be interesting to explore if the effects on proliferation are also reversed in these cells.
Taken together, these findings highlight RIOK2 as a direct link between telomere shortening and anemia associated with aging and related hematological disorders. The bifunctional roles of RIOK2 as a kinase and a transcription factor underscore its extensive regulatory influence over erythropoiesis, with effects on cytoplasmic translation, gene expressions of key hematopoietic transcription factors and telomere homeostasis. This newly described role of RIOK2 as an upstream regulator of the less studied components of telomerase complex (TRiC and dyskerin) also shifts attention towards the importance of the transcriptional regulation of these complexes to maintain telomerase activity. This not only addresses a gap in the current understanding of telomere biology, but also opens a novel perspective on aging and related hematological disorders.
Future research might explore in vivo models to confirm the effects of RIOK2 depletion on telomere length and genomic stability in multicellular organisms, clarifying its therapeutic potential. However, it is important to note that these models may not accurately reflect human conditions due to the naturally varying telomere lengths or telomerase activity throughout lifespan in humans, mice, and zebrafish [61-63]. Another important challenge is the generation of a viable in vivo knockout model for RIOK2. Constitutive knockout of Riok2 results in embryonic lethality in mice [46]. Recently, Messling et al. generated a conditional knockout mouse model (Riok2fl/fl; Rosa26::CreERT2) to assess the effects of whole-body knockout of RIOK2, bypassing the developmental effects [64]. These mice had a median survival of 14.5 days. Additionally, they created a mouse model to assess the loss of Riok2 specifically in the hematopoietic system using Mx1::Cre, which exhibited a median survival of 23.5 days [64]. Thus, constitutive knockout of Riok2 not only leads to embryonic lethality but also results in rapid death in adult mice following its complete loss. This highlights the indispensable role of Riok2 in maintaining healthy hematopoiesis and viability. Therefore, heterozygous depletion of Riok2, either ubiquitously or in a tissue-specific manner, is recommended for in vivo studies.
Another promising avenue is investigating RIOK2’s role in conditions that typically influence telomere dynamics, like oxidative stress or chronic inflammation [65]. These factors are commonly implicated in aging and age-related diseases and could provide further insights into RIOK2’s role in cellular stress responses. Further investigation of RIOK2’s telomere maintenance role could also inform treatments for other conditions associated with shortened telomeres, such as risk of dementia [66], metabolic diseases [67], osteoarthritis [68], and infertility [69]. The causality between telomere shortening and pathogenesis, the molecular causes of altered telomere homeostasis, and the specific cells involved remain areas that require further investigation in these diseases. Correlation analyses of RIOK2 expression levels and telomere length in patient cohorts with age-related diseases, such as cardiovascular disorders and neurodegenerative conditions could also establish its potential as a biomarker. In addition, prospective longitudinal studies could examine whether RIOK2-associated telomere shortening precedes disease onset and/or progression in these conditions, thereby providing a potential screening tool.
Whether pharmacological or genetic modulation of RIOK2 activity can rescue telomere dysfunction in disease settings is another intriguing area of research, that could provide preclinical evidence for its therapeutic application. A major challenge in targeting RIOK2 is its ubiquitous expression in human tissues and indispensable roles in protein synthesis and cell survival. Disrupting these functions in healthy cells could have widespread and unprecedented effects on cellular homeostasis and survival. To mitigate this challenge, tissue or cell-specific depletion of RIOK2 could be adopted. In addition, proteomic screens could identify interacting partners of RIOK2 that could influence its transcriptional functions in telomere maintenance, thus paving ways to uncover novel therapeutic targets. Additionally, epigenetic profiling of RIOK2 promoter and enhancer regions could determine whether distinct modifications influence its activity under stress conditions, such as telomere shortening.
Finally, it is worth noting that RIOK2’s expression levels correlate with prognosis in certain cancers, including tongue squamous cell carcinoma [44]. Cancer cells employ various mechanisms to elongate their telomeres and achieve replicative immortality, such as reactivation of telomerase or the alternative lengthening of telomeres (ALT), a telomerase-independent mechanism mediated by homologous recombination [70,71]. Loss of RIOK2 has been shown to have a more pronounced impact on the proliferation of telomerase-expressing cells than on ALT cells, suggesting that its primary contribution to proliferation defects stems from its role in regulating telomerase activity [47]. However, RIOK2’s loss also dampens ALT cell proliferation, implicating the roles of RIOK2’s downstream targets beyond telomerase maintenance. Investigating RIOK2’s role(s) in cancer progression and exploring it as a therapeutic target could thus be valuable. As a hematological malignancy reliant on telomerase activity, MDS represents one of the most promising diseases for developing treatment strategies by regulating RIOK2.
In conclusion, RIOK2’s newly discovered functions make it a regulatory hub linking transcriptional regulation of erythropoiesis with telomere maintenance (Figure 1), and a plausible target for therapeutic approaches in hematological diseases. Its distinct roles in telomere maintenance also makes it a promising target for aging and other age-related disorders.
References
2. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019 May;20(5):299-309.
3. Pickett HA, Henson JD, Au AY, Neumann AA, Reddel RR. Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum Mol Genet. 2011 Dec 1;20(23):4684-92.
4. Blackburn EH, Collins K. Telomerase: an RNP enzyme synthesizes DNA. Cold Spring Harb Perspect Biol. 2011 May 1;3(5):a003558.
5. Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014 Oct 30;124(18):2775-83.
6. van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997 Feb 20;385(6618):740-3.
7. Smogorzewska A, van Steensel B, Bianchi A, Oelmann S, Schaefer MR, Schnapp G, et al. Control of human telomere length by TRF1 and TRF2. Mol Cell Biol. 2000 Mar;20(5):1659-68.
8. Rivera T, Haggblom C, Cosconati S, Karlseder J. A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol. 2017 Jan;24(1):30-39.
9. McNally EJ, Luncsford PJ, Armanios M. Long telomeres and cancer risk: the price of cellular immortality. J Clin Invest. 2019 Aug 5;129(9):3474-81.
10. Roka K, Solomou EE, Kattamis A. Telomere biology: from disorders to hematological diseases. Front Oncol. 2023 May 19;13:1167848.
11. Revy P, Kannengiesser C, Bertuch AA. Genetics of human telomere biology disorders. Nat Rev Genet. 2023 Feb;24(2):86-108.
12. Batista LFZ, Dokal I, Parker R. Telomere biology disorders: time for moving towards the clinic? Trends Mol Med. 2022 Oct;28(10):882-91.
13. Armanios M. Telomeres and age-related disease: how telomere biology informs clinical paradigms. J Clin Invest. 2013 Mar;123(3):996-1002.
14. Kam MLW, Nguyen TTT, Ngeow JYY. Telomere biology disorders. NPJ Genom Med. 2021 May 28;6(1):36.
15. Silhan LL, Shah PD, Chambers DC, Snyder LD, Riise GC, Wagner CL, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J. 2014 Jul;44(1):178-87.
16. Rocha V, Devergie A, Socié G, Ribaud P, Espérou H, Parquet N, et al. Unusual complications after bone marrow transplantation for dyskeratosis congenita. Br J Haematol. 1998 Oct;103(1):243-8.
17. Cazzola M. Myelodysplastic Syndromes. N Engl J Med. 2020 Oct 1;383(14):1358-74.
18. Syed K, Naguib S, Liu ZJ, Cimmino L, Yang FC. Novel combinations to improve hematopoiesis in myelodysplastic syndrome. Stem Cell Res Ther. 2020 Mar 20;11(1):132.
19. Hellström-Lindberg E, Tobiasson M, Greenberg P. Myelodysplastic syndromes: moving towards personalized management. Haematologica. 2020 Jul;105(7):1765-79.
20. Garcia-Manero G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023 Aug;98(8):1307-25.
21. Sekeres MA, Taylor J. Diagnosis and Treatment of Myelodysplastic Syndromes: A Review. JAMA. 2022 Sep 6;328(9):872-80.
22. Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022 Jul;1(7):EVIDoa2200008.
23. Brunner AM, Leitch HA, van de Loosdrecht AA, Bonadies N. Management of patients with lower-risk myelodysplastic syndromes. Blood Cancer J. 2022 Dec 14;12(12):166.
24. Platzbecker U. Treatment of MDS. Blood. 2019 Mar 7;133(10):1096-107.
25. Rollison DE, Epling-Burnette PK, Park JY, Lee JH, Park H, Jonathan K, et al. Telomere length in myelodysplastic syndromes. Leuk Lymphoma. 2011 Aug;52(8):1528-36.
26. Shin DY, Lim KM, Park HS, Kwon S, Yoon SS, Lee DS. The importance of critically short telomere in myelodysplastic syndrome. Biomark Res. 2022 Nov 10;10(1):79.
27. Myllymäki M, Redd R, Reilly CR, Saber W, Spellman SR, Gibson CJ, et al. Short telomere length predicts nonrelapse mortality after stem cell transplantation for myelodysplastic syndrome. Blood. 2020 Dec 24;136(26):3070-81.
28. Boultwood J, Fidler C, Kusec R, Rack K, Elliott PJ, Atoyebi O, et al. Telomere length in myelodysplastic syndromes. Am J Hematol. 1997 Dec;56(4):266-71.
29. Göhring G, Lange K, Hofmann W, Nielsen KV, Hellström-Lindberg E, Roy L, et al. Telomere shortening, clonal evolution and disease progression in myelodysplastic syndrome patients with 5q deletion treated with lenalidomide. Leukemia. 2012 Feb;26(2):356-8.
30. Reilly CR, Myllymäki M, Redd R, Padmanaban S, Karunakaran D, Tesmer V, et al. The clinical and functional effects of TERT variants in myelodysplastic syndrome. Blood. 2021 Sep 9;138(10):898-911.
31. Yamaguchi H, Baerlocher GM, Lansdorp PM, Chanock SJ, Nunez O, Sloand E, et al. Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood. 2003 Aug 1;102(3):916-8.
32. Nagpal N, Wang J, Zeng J, Lo E, Moon DH, Luk K, et al. Small-Molecule PAPD5 Inhibitors Restore Telomerase Activity in Patient Stem Cells. Cell Stem Cell. 2020 Jun 4;26(6):896-909.e8.
33. Ivancich M, Schrank Z, Wojdyla L, Leviskas B, Kuckovic A, Sanjali A, et al. Treating Cancer by Targeting Telomeres and Telomerase. Antioxidants (Basel). 2017 Feb 19;6(1):15.
34. Röth A, Harley CB, Baerlocher GM. Imetelstat (GRN163L)--telomerase-based cancer therapy. Recent Results Cancer Res. 2010;184:221-34.
35. Keam SJ. Imetelstat: First Approval. Drugs. 2024 Sep;84(9):1149-55.
36. FDA approves imetelstat for low- to intermediate-1 risk myelodysplastic syndromes with transfusion-dependent anemia Accessed Nov 13, 2024]; Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-imetelstat-low-intermediate-1-risk-myelodysplastic-syndromes-transfusion-dependent.
37. Platzbecker U, Santini V, Fenaux P, Sekeres MA, Savona MR, Madanat YF, et al. Imetelstat in patients with lower-risk myelodysplastic syndromes who have relapsed or are refractory to erythropoiesis-stimulating agents (IMerge): a multinational, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2024 Jan 20;403(10423):249-60.
38. Poloni A, Serrani F, Berardinelli E, Maurizi G, Mariani M, Costantini B, et al. Telomere length, c-myc and mad-1 expression could represent prognosis markers of myelodysplastic syndrome. Leuk Res. 2013 Nov;37(11):1538-44.
39. Ohyashiki JH, Iwama H, Yahata N, Ando K, Hayashi S, Shay JW, et al. Telomere stability is frequently impaired in high-risk groups of patients with myelodysplastic syndromes. Clin Cancer Res. 1999 May;5(5):1155-60.
40. Dong W, Qian Y, Yang L. Telomerase, hTERT and splice variants in patients with myelodysplastic syndromes. Leuk Res. 2014 Jul;38(7):830-5.
41. Steensma DP, Fenaux P, Van Eygen K, Raza A, Santini V, Germing U, et al. Imetelstat Achieves Meaningful and Durable Transfusion Independence in High Transfusion-Burden Patients With Lower-Risk Myelodysplastic Syndromes in a Phase II Study. J Clin Oncol. 2021 Jan 1;39(1):48-56.
42. Widmann B, Wandrey F, Badertscher L, Wyler E, Pfannstiel J, Zemp I, et al. The kinase activity of human Rio1 is required for final steps of cytoplasmic maturation of 40S subunits. Mol Biol Cell. 2012 Jan;23(1):22-35.
43. RIOK2. The Human Protein Atlas December 27, 2024]; Available from: https://v16.proteinatlas.org/ENSG00000058729-RIOK2/tissue.
44. Matsuzaki Y, Naito Y, Miura N, Mori T, Watabe Y, Yoshimoto S, et al. RIOK2 Contributes to Cell Growth and Protein Synthesis in Human Oral Squamous Cell Carcinoma. Curr Oncol. 2022 Dec 26;30(1):381-91.
45. Ghosh S, Raundhal M, Myers SA, Carr SA, Chen X, Petsko GA, et al. Identification of RIOK2 as a master regulator of human blood cell development. Nat Immunol. 2022 Jan;23(1):109-21.
46. Raundhal M, Ghosh S, Myers SA, Cuoco MS, Singer M, Carr SA, et al. Blockade of IL-22 signaling reverses erythroid dysfunction in stress-induced anemias. Nat Immunol. 2021 Apr;22(4):520-9.
47. Ghosh S, Nguyen MT, Choi HE, Stahl M, Kühn AL, Van der Auwera S, et al. RIOK2 transcriptionally regulates TRiC and dyskerin complexes to prevent telomere shortening. Nat Commun. 2024 Aug 20;15(1):7138.
48. Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, et al. Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell. 2014 Dec 4;159(6):1389-403.
49. Venteicher AS, Abreu EB, Meng Z, McCann KE, Terns RM, Veenstra TD, et al. A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science. 2009 Jan 30;323(5914):644-8.
50. Zhong F, Savage SA, Shkreli M, Giri N, Jessop L, Myers T, et al. Disruption of telomerase trafficking by TCAB1 mutation causes dyskeratosis congenita. Genes Dev. 2011 Jan 1;25(1):11-6.
51. Chen L, Roake CM, Freund A, Batista PJ, Tian S, Yin YA, et al. An Activity Switch in Human Telomerase Based on RNA Conformation and Shaped by TCAB1. Cell. 2018 Jun 28;174(1):218-230.e13.
52. Mitchell JR, Wood E, Collins K. A telomerase component is defective in the human disease dyskeratosis congenita. Nature. 1999 Dec 2;402(6761):551-5.
53. Shukla S, Jeong HC, Sturgeon CM, Parker R, Batista LFZ. Chemical inhibition of PAPD5/7 rescues telomerase function and hematopoiesis in dyskeratosis congenita. Blood Adv. 2020 Jun 23;4(12):2717-22.
54. MacNeil DE, Lambert-Lanteigne P, Autexier C. N-terminal residues of human dyskerin are required for interactions with telomerase RNA that prevent RNA degradation. Nucleic Acids Res. 2019 Jun 4;47(10):5368-80.
55. Parry EM, Alder JK, Lee SS, Phillips JA 3rd, Loyd JE, Duggal P, et al. Decreased dyskerin levels as a mechanism of telomere shortening in X-linked dyskeratosis congenita. J Med Genet. 2011 May;48(5):327-33.
56. Yuan X, Larsson C, Xu D. Mechanisms underlying the activation of TERT transcription and telomerase activity in human cancer: old actors and new players. Oncogene. 2019 Aug;38(34):6172-83.
57. Dogan F, Forsyth NR. Telomerase Regulation: A Role for Epigenetics. Cancers (Basel). 2021 Mar 10;13(6):1213.
58. Ramos C, Montaño M, García-Alvarez J, Ruiz V, Uhal BD, Selman M, et al. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001 May;24(5):591-8.
59. Álvarez D, Cárdenes N, Sellarés J, Bueno M, Corey C, Hanumanthu VS, et al. IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol. 2017 Dec 1;313(6):L1164-73.
60. Pechkovsky DV, Prêle CM, Wong J, Hogaboam CM, McAnulty RJ, Laurent GJ, et al. STAT3-mediated signaling dysregulates lung fibroblast-myofibroblast activation and differentiation in UIP/IPF. Am J Pathol. 2012 Apr;180(4):1398-412.
61. Kipling D, Cooke HJ. Hypervariable ultra-long telomeres in mice. Nature. 1990 Sep 27;347(6291):400-2.
62. Anchelin M, Murcia L, Alcaraz-Pérez F, García-Navarro EM, Cayuela ML. Behaviour of telomere and telomerase during aging and regeneration in zebrafish. PLoS One. 2011 Feb 9;6(2):e16955.
63. Lund TC, Glass TJ, Tolar J, Blazar BR. Expression of telomerase and telomere length are unaffected by either age or limb regeneration in Danio rerio. PLoS One. 2009 Nov 6;4(11):e7688.
64. Messling JE, Peña-Rømer I, Moroni AS, Bruestl S, Helin K. RIO-kinase 2 is essential for hematopoiesis. PLoS One. 2024 Apr 2;19(4):e0300623.
65. Schellnegger M, Hofmann E, Carnieletto M, Kamolz LP. Unlocking longevity: the role of telomeres and its targeting interventions. Front Aging. 2024 Jan 25;5:1339317.
66. Cao Z, Hou Y, Xu C. Leucocyte telomere length, brain volume and risk of dementia: a prospective cohort study. Gen Psychiatr. 2023 Sep 11;36(4):e101120.
67. Gao Z, Daquinag AC, Fussell C, Zhao Z, Dai Y, Rivera A, et al. Age-associated telomere attrition in adipocyte progenitors predisposes to metabolic disease. Nat Metab. 2020 Dec;2(12):1482-97.
68. Poonpet T, Saetan N, Tanavalee A, Wilairatana V, Yuktanandana P, Honsawek S. Association between leukocyte telomere length and angiogenic cytokines in knee osteoarthritis. Int J Rheum Dis. 2018 Jan;21(1):118-25.
69. Xu X, Chen X, Zhang X, Liu Y, Wang Z, Wang P, et al. Impaired telomere length and telomerase activity in peripheral blood leukocytes and granulosa cells in patients with biochemical primary ovarian insufficiency. Hum Reprod. 2017 Jan;32(1):201-7.
70. Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017 Mar;49(3):349-57.
71. Zhang JM, Zou L. Alternative lengthening of telomeres: from molecular mechanisms to therapeutic outlooks. Cell Biosci. 2020 Mar 10;10:30.