Abstract
Allogeneic hematopoietic stem cell transplantation is often the preferred treatment for patients with hematopoietic malignancies, affording them the best chance for long-term disease-free survival. Because related donors are the preferred source of hematopoietic stem/progenitor cells, consideration of familial predisposition to cancer should become standardized as part of pre-transplant assessments. Based on numerous published cases of poor outcomes for both the donor and recipient when allogeneic donors with deleterious germline variants in RUNX1 or CEBPA have been used for hematopoietic stem cell transplantation, such donors should be avoided. However, data do not exist for many other genes, which presents challenges to providers who are counseling patients and potential donors about risk. Hopefully, ongoing studies will fill these gaps and inform recommendations regarding suitability of universal donor screening in the future.
Case Presentation
A 56-year-old Caucasian man was referred to an academic medical center for consideration of allogeneic hematopoietic stem cell transplantation (HSCT). One year earlier, he had seen his primary care physician for increased fatigue and decreased exercise tolerance, and a complete blood cell count showed a total white blood cell count of 2500/μL, hemoglobin of 7.2g/dL, and a platelet count of 110,000/μL. The white blood cell differential showed an absolute neutrophil count of 900/μL. The patient was referred to a hematologist who performed a bone marrow biopsy that showed a myelodysplastic syndrome with del(5q). Molecular profiling showed a deleterious DNMT3A variant at a variant allele frequency (VAF) of 35%, and two DDX41 variants: p.Asp140Gfs*2 [hereafter abbreviated as D140fs] at VAF 49% and R525H at 34%. The patient was treated with monthly cycles of 5-azacytidine with rapid improvement in his peripheral blood cell counts, and a referral for allogeneic HSCT was made. Because the DDX41 D140fs allele is a recurring germline variant in Caucasian populations, a skin biopsy was performed, and the D140fs allele was observed in DNA derived from cultured skin fibroblasts, confirming its germline status. Cascade testing throughout the family was initiated, and the allogeneic HSCT was performed using an unrelated HSCT donor. The patient did well during HSCT and was discharged with recovering hematopoiesis at day +14. At day +30, a standard bone marrow biopsy was performed. Microsatellite markers were used to test donor chimerism, which was at >95%, and molecular profiling was performed for measuring residual disease. No variants in DNMT3A or DDX41 were observed, but a BRCA1 185delAG, an Ashkenazi Jewish founder mutation, was demonstrated at a VAF of 45%.
This case raises several important issues, each of which is discussed in more detail below:
- When should transplant teams think about germline predisposition to hematopoietic malignancies?
- How should molecular profiling data from the patient’s hematopoietic malignancy be interpreted relative to germline status of variants?
- Should hematopoietic stem cells from related donors with deleterious germline variants be used for allogeneic HSCT?
- What should transplant teams do if a deleterious variant that predisposes to cancer is found in donor cells post-transplant?
- Should all allogeneic HSCT donors be screened for deleterious germline predisposition variants?
- Should preemptive allogeneic stem cell transplantation be considered for carriers of deleterious germline variants in genes that predispose to hematopoietic malignancies?
When should transplant teams think about germline predisposition to hematopoietic malignancies?
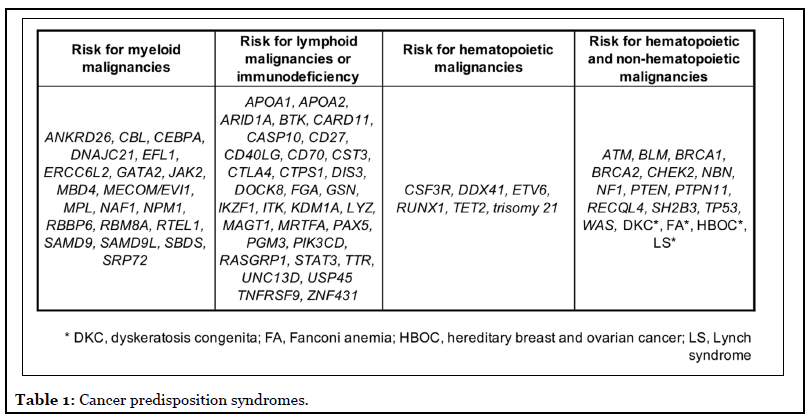
Simply put, transplant teams should think about germline predisposition to hematopoietic malignancies for every patient being considered for transplant. The sheer number of germline predisposition syndromes (Table 1) [1-8] makes this a challenging task for most clinicians. During transplantation, a relatively small number of hematopoietic stem and progenitor cells (HSPCs) is implanted into the recipient, regardless of whether the HSPCs are from the recipient her-/himself (autologous) or from another individual (allogeneic). These HSPCs are under replicative stress to reconstitute a bone marrow in the recipient, and they will contain any germline variants that were present in the individual from whom they were collected. If those germline variants are deleterious and in genes encoding master transcription factors RUNX1 or CEBPA, several poor outcomes have been noted: poor donor mobilization [9,10], failure or delay in engraftment [9,11], poor immune function post-transplant [9,11], early relapse [11], donorderived leukemias in transplant recipients [10,12,13], and leukemia development in the related donor after stem cell mobilization/collection [12,13], all suggesting that use of related donors with deleterious germline mutations in these two genes is ill-advised. We have much less data on HSPCs with deleterious germline variants in other cancerpredisposing genes, and additional data are needed to determine if there are any germline variants that are permissive to HSCT. When relatives are used as donors, their personal and family histories are more accessible to the transplant team. Unfortunately, much less information about personal/family history is provided for unrelated donors or umbilical cord units, but of course, these HSPC sources will contain germline variants as well.
For HSCT recipients found to have a deleterious germline variant that predisposes to cancer, it is important to remember that all non-germ cells contain that person’s germline variants. Therefore, post-transplant, regardless of which donor is used, the recipient’s germline variants remain in the bone marrow within the non-hematopoietic mesenchymal stromal cells as well as in other tissues. Whether the presence of the germline variant within these cells has an impact on the outcome of the HSCT is not known for most cancer predisposition genes. To date, the most data for outcomes of HSCT in patients with deleterious germline variants are most robust for those with disease-causing germline GATA2 variants.
Patients with deleterious germline GATA2 mutations often have deficiencies in monocytes, B cells, natural killer cells, and dendritic cells, leading to infections by fungal, viral, and bacterial species, especially non-tuberculous Mycobacteria. Because of the recognized immunodeficiency of these individuals and their life-long risk of infection, investigators at the National Institutes of Health have optimized allogeneic HSCT for these individuals [14-16]. With a median follow-up of two years, 19 of 22 patients (86%) were alive with correction of phenotypes associated with GATA2-deficiency, clearance of dysplasia, and normalization of karyotypic abnormalities. Both acute and chronic forms of graft versus host disease were the main toxicities observed.
HSCT outcomes have also been assessed in pediatric patients with GATA2 deficiency [17]. Investigators compared the outcomes of young individuals with diseasecausing GATA2 variants who had developed bone marrow failure (BMF), myelodysplastic syndrome (MDS), or acute myeloid leukemia (AML) compared to those without such variants in a retrospective case-control study. Although the 5-year overall and disease-free survival rates were similar in the patient cohorts, the 5-year event-free survival rate was lower in those with deleterious GATA2 variants: 7% ± 6% versus 28-33% ± 10% in controls. Treatmentrelated toxicities, such as neurologic toxicities, infectious complications, and thromboses post-transplant, were responsible for this difference. Among four people with neurologic toxicities, severe peripheral polyneuropathy was observed in three individuals, noninfectious encephalopathy in one, and unexplained mental status changes/delirium in two. The investigators found no differences in treatment related mortality or graft-versushost disease among the groups.
Recently, two studies have examined the HSCT outcomes of children with deleterious germline SAMD9/SAMD9L variants. Sarthy et al. published a small series of two cases of patients with pathogenic germline SAMD9 variants and MIRAGE syndrome, a constellation of problems including infection, growth restriction, adrenal hypoplasia, genital abnormalities, enteropathy, and high risk for development of MDS, who underwent HSCT, one using an HLAmismatched unrelated donor and the other using a matched related donor [18]. Both of these patients died shortly after transplant due to complications stemming from their co-morbid conditions, including temperature instability, enteropathy, adrenal insufficiency, electrolyte disorders, susceptibility to infection, and lung disease.
Better outcomes were seen in more recent experience with a 12-patient cohort of patients with deleterious germline variants in either SAMD9 or SAMD9L all of whom underwent allogeneic HSCT for MDS (ten individuals), congenital amegakaryocytic thrombocytopenia (one patient), and dyskeratosis congenita (one patient) [19]. The median age at HCT was 2.8 years (range, 1.2 to 12.8 years), and myeloablative conditioning was used in nine children, with reduced intensity conditioning used in the others. One patient with a pathogenic SAMD9L variants failed to engraft and died of refractory myeloid malignancy, and another who engrafted died of diffuse alveolar hemorrhage. Among the other ten patients who engrafted successfully, the immediate post-transplant course was complicated by underlying co-morbidities for those with MIRAGE syndrome, but had resolution of their hematopoietic malignancies and achieved sustained donor engraftment. Overall, 10/12 patients were alive with a median follow-up of 3.1 years (range, 0.1 to 14.7 years). Taken together, these publications suggest that specific protocols may need to be developed to optimize HSCTs in these individuals with very specific co-morbidities that arise from the germline syndrome.
How should molecular profiling data from the patient’s hematopoietic malignancy be interpreted relative to germline status of variants?
Because germline variants are present in all non-germ cells of an individual’s body, malignant cells will have that person’s germline variants. Therefore, when molecular profiling is performed on DNA derived from malignant cells, observed variants can be somatic or germline in nature [20]. When such molecular profiling is done sequentially over time as a patient’s disease status changes, variants that remain at a consistently high VAF are often of germline origin, especially when there has been a change in the patient’s disease status, for example from leukemia present to remission. In some situations when eradication of the somatic mutation is not expected, for example for a patient with a chronic myeloproliferative disease, persistence of a JAK2 V617F variant most likely represents the underlying malignancy, rather than a germline allele. However, in many cases, persistence of a deleterious variant can be used to prioritize an individual for germline testing. This approach should not be used in place of proper germline testing, since most panels used for prognostication do not contain all of the genes that confer germline risk for cancer and/or are often not capable of detecting deletions or other copy number variants that commonly cause germline predisposition. Therefore, if the personal or family history suggest germline predisposition, proper germline testing using true germline DNA and a testing platform comprehensive for the relevant genes/mutation types should be undertaken. Elements of the personal history that should raise concern for germline predisposition include: diagnosis of a malignancy at a unusually early age compared to the general population, e.g., MDS at <40 years old; history of multiple malignancies; and/or a physical examination that reveals features consistent with a germline predisposition syndrome [21]. Elements of the family history that signal the need for germline genetic risk testing include: history of bleeding, cytopenias, and/ or the presence of a hematopoietic or young-onset (<50 years) solid tumor within two generations of the proband. As noted above, review of molecular profiling of the patient’s malignant cells is another means of identifying individuals who may have germline cancer predisposition alleles. As the opening case shows, when these data are available, they can alert astute clinicians to patients with germline variants. It is important to note that not all germline variants are deleterious, and classification of germline variants should be done in accordance with the recommendations of the American College of Medical Genetics and Genomics [22].
Should hematopoietic stem cells from related donors with deleterious germline variants be used for allogeneic HSCT?
Unfortunately, published standards for the evaluation of related donors do not include evaluation of germline cancer predisposition syndromes [23,24]. Phenotypes observed in donors that should signal concern for germline predisposition and prompt a thorough workup include baseline thrombocytopenia [25] and/or poor donor mobilization [10]. Although clonal hematopoiesis (CH) represents somatic mutation within HSPCs and is therefore outside the scope of this discussion, many now advocate avoiding using donors with this condition [10,26-28]. Although studies to date have not seen an effect on overall survival of HSCT recipients when donors with CH have been used, a higher incidence of graft-versus-host disease has been noted, with one study showing more acute GVHD [29], and another more chronic GVHD [30]. Even rare clones in donor cell collections can expand in HSCT recipients [31]. Many studies acknowledge the risk of donor-derived leukemias arising in HSCT recipients who have received donors’ cells with CH [10,26-28,30,32-34].
Unfortunately, data do not exist on the outcomes of donors with deleterious germline variants who underwent growth-factor mobilization and HSCT recipients who were transplanted with HSPCs containing such variants. However as noted above, retrospective studies of the outcomes of HSCTs that have used donor cells with deleterious variants in RUNX1 and CEBPA demonstrate devastating consequences: poor HSC mobilization by the donor [9,10], failure or delay in engraftment [9,11], poor immune function post-transplant [9,11], early relapse [11], donor-derived leukemias in transplant recipients [10,12,13], and leukemia development in the related donor after stem cell mobilization/collection [12,13].
For these reasons, the author advocates a careful review of personal and family histories and molecular testing performed to characterize hematopoietic malignancies at the time of consideration of allogeneic HSCT using related donors. Cascade testing of potential related donors is best performed at the presentation of the hematopoietic malignancy since germline genetic testing takes time, and this allows maximal discussion with the patient and potential donor(s). Whenever possible, we avoid using related donors with known deleterious germline variants to decrease the risk of poor engraftment, graft dysfunction/ failure, or subsequent donor-derived leukemias, although we recognize that data are limited outside of RUNX1 and CEBPA. These discussions require delicacy in balancing protection of health information and patient confidentiality with informing individuals adequately of their risks.
What should transplant teams do if a deleterious variant that predisposes to cancer is found in donor cells post-transplant?
As molecular profiling is being used for measuring residual disease post-transplant, germline variants are being detected in unrelated donor cells, as the opening case demonstrates. These situations present very difficult ethical scenarios both in terms of how to discuss such findings and their implications with the HSCT recipient as well as whether it is appropriate to disclose such information back to the donor center and eventually to the donor. The author anticipates that this scenario will be recognized increasingly as molecular profiling is instituted broadly and expands to include all of the known cancerpredisposing genes. At the moment, the author advocates offering disclosure of this information to HSCT recipients with appropriate genetic counseling as well as to the donor center that provided the HSPCs.
Should all allogeneic HSCT donors be screened for deleterious germline predisposition variants?
All people have several deleterious germline variants in their genomes, most of which are recessive. Therefore, it should be no surprise that all donor cells have such variants, whether the cells come from relatives or from more anonymous sources. As the number of cancerpredisposing genes expands, with many of these syndromes recognized as dominant, we may soon come to the point of recommending that all donor sources are screened for deleterious germline predisposition variants. We are likely to need more data as to which variants are permissive to HSCT and which, like those in RUNX1 and CEBPA, present barriers to successful HSCT. Once these data are available and confident predictions can be made regarding the impact of particular germline variants, then it may be advantageous to do universal germline genetic screening of all donors.
Should preemptive allogeneic stem cell transplantation be considered for carriers of deleterious germline variants in genes that predispose to hematopoietic malignancies?
There may be some scenarios when physicians should consider preemptive allogeneic HSCT prior to the development of malignancy. Two that are worth discussion are: (i) for highly penetrant germline genetic syndromes. For example, individuals with deleterious variants at the 5’ end of CEBPA are virtually guaranteed of developing a myeloid malignancy in their lifetime. As such, some have argued that the risk of allogeneic HSCT is less than that of developing malignancy, and preemptive allogeneic HSCT is reasonable [35]. (ii) for those with GATA2-deficiency and a life-long risk of immunodeficiency and infection [36].
Final Thoughts
The increased recognition of the importance of germline cancer-predisposing syndromes and the use of nextgeneration sequencing platforms to diagnose these conditions and to profile hematopoietic malignancies presents challenges to consideration of HSCT. Much of the uncertainty surrounding issues of HSCT recipient/ donor evaluations and counseling derive from a lack of data. The author hopes that the coming years will provide clarity regarding the frequency of germline predisposition in certain diagnoses; the impact of deleterious germline variants in HSCT recipients and donors; and the potential for universal donor screening for these conditions as a way to improve outcomes for patients, their family members, and people generous enough to donate HSPCs.
References
2. Wiggins M, Stevenson W. Genetic predisposition in acute leukaemia. International Journal of Laboratory Hematology. 2020 Mar 2.1:75-81.
3. Carraway H, LaFramboise T. Myeloid neoplasms with germline predisposition: Practical considerations and complications in the search for new susceptibility loci. Best Practice & Research Clinical Haematology. 2020 Jun 8:101191.
4. Rafei H, DiNardo CD. Hereditary myeloid malignancies. Best Practice & Research Clinical Haematology. 2019 Jun 1;32(2):163-76.
5. Galera P, Dulau-Florea A, Calvo KR. Inherited thrombocytopenia and platelet disorders with germline predisposition to myeloid neoplasia. International Journal of Laboratory Hematology. 2019 May;41:131-41.
6. Kallen ME, Dulau-Florea A, Wang W, Calvo KR.Acquired and germline predisposition to bone marrow failure: diagnostic features and clinical implications.InSeminars in Hematology 2019 Jan 1 (Vol. 56, No. 1, pp.69-82). WB Saunders.
7. Weinberg OK, Kuo F, Calvo KR. Germline Predisposition to Hematolymphoid Neoplasia: 2017 Society for Hematopathology/European Association for Haematopathology Workshop Report. American Journal of Clinical Pathology. 2019 Aug 1;152(3):258-76.
8. Bloom M, Maciaszek JL, Clark ME, Pui CH, Nichols KE. Recent advances in genetic predisposition to pediatric acute lymphoblastic leukemia. Expert Review of Hematology. 2020 Jan 2;13(1):55-70.
9. Fogarty PF, Yamaguchi H, Wiestner A, Baerlocher GM, Sloand E, Zeng WS, et al. Late presentation of dyskeratosis congenita as apparently acquired aplastic anaemia due to mutations in telomerase RNA. The Lancet. 2003 Nov 15;362(9396):1628-30.
10. Rojek K, Nickels E, Neistadt B, Marquez R, Wickrema A, Artz A, et al. Identifying inherited and acquired genetic factors involved in poor stem cell mobilization and donor-derived malignancy. Biology of Blood and Marrow Transplantation. 2016 Nov 1;22(11):2100-3.
11. Owen CJ, Toze CL, Koochin A, Forrest DL, Smith CA, Stevens JM, et al. Five new pedigrees with inherited RUNX1 mutations causing familial platelet disorder with propensity to myeloid malignancy. Blood, The Journal of the American Society of Hematology. 2008 Dec 1;112(12):4639-45.
12. Buijs A, Poddighe P, van Wijk R, van Solinge W, Borst E, Verdonck L, et al. A novel CBFA2 single-nucleotide mutation in familial platelet disorder with propensity to develop myeloid malignancies. Blood, The Journal of the American Society of Hematology. 2001 Nov 1;98(9):2856- 8.
13. Xiao H, Shi J, Luo Y, Tan Y, He J, Xie W, et al. First report of multiple CEBPA mutations contributing to donor origin of leukemia relapse after allogeneic hematopoietic stem cell transplantation. Blood, The Journal of the American Society of Hematology. 2011 May 12;117(19):5257-60.
14. Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, Hsu AP, Zerbe CS, Calvo KR, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood. 2011 Sep 29;118(13):3715-20.
15. Grossman J, Cuellar-Rodriguez J, Gea-Banacloche J, Zerbe C, Calvo K, Hughes T, et al. Nonmyeloablative allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Biology of Blood and Marrow Transplantation. 2014 Dec 1;20(12):1940-8.
16. Parta M, Shah NN, Baird K, Rafei H, Calvo KR, Hughes T, et al. Allogeneic hematopoietic stem cell transplantation for GATA2 deficiency using a busulfan-based regimen. Biology of Blood and Marrow Transplantation. 2018 Jun 1;24(6):1250-9.
17. Hofmann I, Avagyan S, Stetson A, Guo D, Al- Sayegh H, London WB, et al. Comparison of Outcomes of Myeloablative Allogeneic Stem Cell Transplantation for Pediatric Patients with Bone Marrow Failure, Myelodysplastic Syndrome and Acute Myeloid Leukemia with and without Germline GATA2 Mutations. Biology of Blood and Marrow Transplantation. 2020 Feb 20:1124-30.
18. Sarthy J, Zha J, Babushok D, Shenoy A, Fan JM, Wertheim G, et al. Poor outcome with hematopoietic stem cell transplantation for bone marrow failure and MDS with severe MIRAGE syndrome phenotype. Blood Advances. 2018 Jan 23;2(2):120-5.
19. Ahmed IA, Farooqi MS, Vander Lugt MT, Boklan J, Rose M, Friehling ED, et al. Outcomes of hematopoietic cell transplantation in patients with germline SAMD9/ SAMD9L mutations. Biology of Blood and Marrow Transplantation. 2019 Nov 1;25(11):2186-96.
20. Drazer MW, Kadri S, Sukhanova M, Patil SA, West AH, Feurstein S, et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Advances. 2018 Jan 23;2(2):146-50.
21. University of Chicago Hematopoietic Malignancies Cancer Risk Team. How I diagnose and manage individuals at risk for inherited myeloid malignancies. Blood, The Journal of the American Society of Hematology. 2016 Oct 6;128(14):1800-13.
22. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine. 2015 May;17(5):405-23.
23. Worel N, Buser A, Greinix HT, Hägglund H, Navarro W, Pulsipher MA, et al. Suitability criteria for adult related donors: a consensus statement from the Worldwide Network for Blood and Marrow Transplantation Standing Committee on Donor Issues. Biology of Blood and Marrow Transplantation. 2015 Dec 1;21(12):2052-60.
24. Churpek JE, Artz A, Bishop M, Liu H, Godley LA. Correspondence regarding the consensus statement from the worldwide network for blood and marrow transplantation standing committee on donor issues. Biology of Blood and Marrow Transplantation. 2016 Jan 1;22(1):183-4.
25. Churpek JE, Nickels E, Marquez R, Rojek K, Liu B, Lorenz R, et al. Identifying familial myelodysplastic/ acute leukemia predisposition syndromes through hematopoietic stem cell transplantation donors with thrombocytopenia. Blood, The Journal of the American Society of Hematology. 2012 Dec 20;120(26):5247-9.
26. Gondek LP, Zheng G, Ghiaur G, DeZern AE, Matsui W, Yegnasubramanian S, et al. Donor cell leukemia arising from clonal hematopoiesis after bone marrow transplantation. Leukemia. 2016 Sep;30(9):1916-20.
27. Burns SS, Kapur R. Clonal Hematopoiesis of Indeterminate Potential as a Novel Risk Factor for Donor-Derived Leukemia. Stem Cell Reports. 2020 Aug 11;15(2):279-91.
28. DeZern AE, Gondek LP. Point: Stem cell donors should be screened for CHIP. Blood Advances. 2020 Feb 25;4(4):784-8.
29. Oran B, Champlin RE, Wang F, Jeyakumar N, Garcia-Manero G, Kantarjian HM, et al. Donor clonal hematopoiesis increases risk of acute graft versus host disease after matched related transplantation in AML and MDS patients. Blood. 2019 Nov 13;134(Supplement_1).
30. Frick M, Chan W, Arends CM, Hablesreiter R, Halik A, Heuser M, et al. Role of donor clonal hematopoiesis in allogeneic hematopoietic stem-cell transplantation. Journal of Clinical Oncology. 2019 Feb 10;37(5):375-85.
31. Wong WH, Bhatt S, Trinkaus K, Pusic I, Elliott K, Mahajan N, et al. Engraftment of rare, pathogenic donor hematopoietic mutations in unrelated hematopoietic stem cell transplantation. Science Translational Medicine. 2020 Jan 15;12(526).
32. Park DS, Akuffo AA, Muench DE, Grimes HL, Epling- Burnette PK, Maini PK, et al. Clonal hematopoiesis of indeterminate potential and its impact on patient trajectories after stem cell transplantation. PLoS Computational Biology. 2019 Apr 26;15(4):e1006913.
33. Boettcher S, Wilk CM, Singer J, Beier F, Burcklen E, Beisel C, et al. Clonal hematopoiesis in donors and longterm survivors of related allogeneic hematopoietic stem cell transplantation. Blood, The Journal of the American Society of Hematology. 2020 Apr 30;135(18):1548-59.
34. Nawas MT, Schetelig J, Damm F, Levine RL, Angel- Perales M, Giralt SA, et al. The clinical implications of clonal hematopoiesis in hematopoietic cell transplantation. Blood Reviews. 2020 Aug 24:100744.
35. Hamilton KV, Maese L, Marron JM, Pulsipher MA, Porter CC, Nichols KE. Stopping leukemia in its tracks: should preemptive hematopoietic stem-cell transplantation be offered to patients at increased genetic risk for acute myeloid leukemia?. Journal of Clinical Oncology. 2019 Aug 20;37(24):2098-104.
36. Bogaert DJ, Laureys G, Naesens L, Mazure D, De Bruyne M, Hsu AP, et al. GATA2 deficiency and haematopoietic stem cell transplantation: challenges for the clinical practitioner. British Journal of Haematology. 2020 Mar;188(5):768-73.