Abstract
The Epstein-Barr Virus-associated T- and NK-cell lymphoproliferative disorders are a group of hematologic disturbances that occur in pediatric age groups. The literature describes extensive heterogeneity in the presentation of this disease, with a noncanonical reaction to Epstein-Barr virus infection underpinning the wide variety of clinical manifestations. In this review, we discuss chronic active Epstein-Barr virus infection, hypersensitivity to mosquito bites, Alice in Wonderland syndrome, Hydroa vacciniforme-like lymphoproliferative disorder, and the clinical and histologic findings of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis and systemic Epstein-Barr-positive T-cell lymphoproliferative disorder of childhood. Several case reports of Epstein-Barr virus-associated hemophagocytic lymphohistiocytosis and systemic Epstein-Barr positive T-cell lymphoproliferative disorder of childhood describe extensive hemophagocytic lymphohistiocytosis, suggesting overlap between the various entities of Epstein-Barr Virus-associated T- and NK-cell lymphoproliferative disorders. We hope to provide a comprehensive overview of several possible manifestations of EBV-associated systemic T-cell lymphoproliferative disorders and demonstrate that extensive overlap occurs between the various entities.
Abbreviations
EBV: Epstein-Barr Virus; EBER: Epstein–Barr Virus-Encoded Small RNAs; HLH: Hemophagocytic Lymphohistiocytosis; DIC: Disseminated Intravascular Coagulopathy
Introduction
The Epstein-Barr Virus (EBV) is an endemic human herpesvirus, with 90% of people demonstrating serological evidence of prior exposure to EBV. In most cases, primary infections of EBV generate mild or asymptomatic clinical courses, but may present as infectious mononucleosis if contracted during young adulthood. In a small percentage of cases, Epstein- Barr virus infection may manifest as a noncanonical reaction, yielding a wide variety of clinical manifestations.
A rare but serious complication of primary EBV infection is the development of an EBV-associated lymphoproliferative complications, which can have devastating outcomes [1]. These entities are generally classified under the larger family of EBV-positive lymphoproliferative disorders [2]. This mini review describes the various lymphoproliferative disorders associated with EBV infection, outlining the pathophysiology, histology, and prognosis of each lymphoproliferative manifestation of EBV infection.
Canonical EBV Exposure
Primary EBV exposure is experienced by over 90% of adults by the age of 25. The virus is cleared in approximately 90% of those exposed to EBV, though approximately 10% of exposed individuals will develop infectious mononucleosis, the most usual form of primary EBV exposure. This number is as high as 50% in the young adolescent age group. Infectious mononucleosis is the consequence of EBV infection of the B-cell lineage, with the sparring of T and NK-cell lineages.
Patients will generally present with fatigue, fever, pharyngitis and cervical lymphadenopathy over the course of several weeks. Appreciable lymphadenopathy will begin immediately, then peak by the second week of infection. The microscopic appearance of tonsillar tissue shows a heterogenous infiltrate of various sized lymphocytes, histiocytes, plasma cells, eosinophils and immunoblasts causing effacement of normal lymph node architecture. Follicular hyperplasia with tingible body macrophages and marked mitotic activity may also be seen, due to the reactive process causing these features. Lymph node tissue sampled from patients with infectious mononucleosis may appear grossly abnormal due to the polyclonal expansion and diffuse infiltration of these often atypical appearing large B-cell immunoblasts. Furthermore, the reactive expansion of an oligoclonal lineage of CD8(+) cytotoxic T-cells may be a consequence of B-cell hyperplasia rather than as a direct manifestation of EBV infection itself. These T-cells may also appear atypical, but this process can be distinguished from a T-cell lymphoma since infected B cells will be positive for EBV-encoded RNA (EBER).
Non-canonical EBV Exposure Resulting in Secondary EBV Infection
Following exposure, primary EBV infection is often cleared, yielding lifelong seropositivity to EBV characterized by the presence of EBV IgG. However, active EBV infection may also progress to a systemic chronic active EBV infection (CAEBV), characterized by symptoms of infectious mononucleosis that have persisted for more than three months, the presence of EBV DNA in the peripheral blood, end-organ damage, and the presence of a positive EBER or EBV protein within affected tissues [3]. Immune system dysfunction is not characteristically associated with CAEBV infection, though proliferating T- and NK-cells may infiltrate the parenchyma of various organs involved [4]. These proliferating cells are not overtly abnormal, but their activation may cause end-organ damage. However, marked immune dysfunction can be seen with rarer but more unusual subtypes of CAEBV.
Hypersensitivity to mosquito bites is another manifestation of secondary EBV infection, characterized by a severe local reaction of tense bullae and necrotizing macules forming at the location of the mosquito bite. This condition has been associated with a 50% mortality rate due to the secondary complications from these large local reactions. Histology of the bite site shows ulcer formation with epidermal necrosis, as well as an infiltrate of polyclonal T-lymphocytes and NK-cells, some of which may be EBER-positive. The T-cells are CD4(+) and CD8(+), potentiating the cytotoxic response that drives this severe reaction. These patients may also demonstrate high serum IgE levels, the presence of EBV DNA in the peripheral blood, and peripheral NK- and T-lymphocytosis [5].
Hydroa vacciniforme-like lymphoproliferative disorder is a subtype of CAEBV manifesting as a characteristic cutaneous photodermatosis. HV-LPD may present as a photosensitive vesicular rash in sun-exposed skin, but more severe forms may develop on unexposed areas. The histology is similar to mosquito bite hypersensitivity reaction, with epidermal degeneration and a polymorphic infiltration of EBV-infected small lymphocytes into all layers of the skin, down to the subcutaneous fat. This inflammation takes on a combined septal and lobular panniculitis pattern, originating from periadnexal small vessels. However, this lymphocytic infiltrate comprises primarily of CD4(+) and CD8(+) T-cells, with few NKcells involved. Depending on severity, patients may experience end-organ damage from the subsequent systemic infiltration of these proliferating T-lymphocytes.
Several case reports have also noted an Alice in Wonderland Syndrome from secondary EBV exposure [6]. First described in 1977, Alice in Wonderland Syndrome is characterized by disturbances to visual perception following prolonged infection by infectious mononucleosis. Patients with Alice in Wonderland Syndrome will experience a visual metamorphosia present with intermittent illusions of size, shape and color, with no underlying central nervous system cause. These patients will have a normal ophthalmologic examination, and often do not present with any other visual disturbances. The underlying pathophysiology of Alice in Wonderland Syndrome is unknown.
EBV-positive T/NK-associated Lymphoproliferative Disorders of Childhood
In rare cases, T and NK-cells will be infected during the primary exposure to EBV, and these infections have been associated with fulminant disease and grave clinical outcomes [7]. These cases generally occur in late childhood or early adulthood. Therefore, EBV-positive T/NK-associated lymphoproliferative disorders of childhood are a rare but aggressive manifestation of primary EBV infection. There are two distinct entities associated with EBV-positive T/NKassociated lymphoproliferative disorders of childhood: EBVpositive hemophagocytic lymphohistiocytosis (EBV-HLH) and systemic EBV-positive T-cell lymphoma of childhood [8], with many shared characteristics but significantly different prognoses [9,10].
Clinical Features
In EBV-HLH, infected CD8(+) T-cells may release proinflammatory cytokines leading to macrophage activation, immune dysregulation and a hyperinflammatory state [11]. A sinusoidal and periportal infiltrate of EBER-positive small T-cells can be seen on liver biopsies, with variable hyperplasia and hemophagocytosis of the hepatic macrophages (Kupffer cells). Marked lymphocytosis and histiocytosis can be seen on bone marrow aspirates, with hemophagocytosis and concomitant hypoplasia of the myeloid and erythroid lineages. EBER-positive small T-cells can also be seen within the bone marrow. Clinically, EBV-HLH is the most common cause of secondary hemophagocytic lymphohistiocytosis, comprising approximately 40% of all cases [12]. EBV-HLH can be effectively controlled with supportive measures in 90% of cases, however approximately 10% of children with EBV-HLH experience terminal complications of fulminant hepatic or myeloid failure. The absence of an overt neoplastic process with clonal T-cell receptor gene rearrangement distinguishes EBV-HLH from systemic EBV-positive T-cell lymphoma of childhood, evaluable on bone marrow evaluation or solid organ biopsy.
Systemic EBV-positive T-cell lymphoma of childhood presents as a rapidly clonal proliferation of EBV-infected CD8(+) T-cells. Patients will generally experience malaise and fever preceding the onset of a nonspecific viral respiratory illness, before experiencing an acute decline. Hepatosplenomegaly, acute liver failure and lymphadenopathy can manifest within a period of days to weeks, with rapid progression from sepsis, multi-organ failure, a concomitant HLH syndrome and death. Pancytopenia, abnormal liver enzymes and abnormal EBV serology are commonly observed laboratory abnormalities in patients with this disease.
Systemic EBV-positive T-cell lymphoma of childhood appears more prevalent in Japan, Chinese Taipei, and China than in the Americas, but several case reports have been published from Mexico, South America, and Canada. In several case series, systemic EBV-positive T-cell lymphoma of childhood had a mortality rate approaching 100% [13] whereas the mortality from EBV-HLH ranges from 58% to 80% [10,14]. There appears to be a dichotomy in the outcome of these cases of systemic EBV-positive T-cell lymphoma of childhood, with a better outcome for the cases occurring in Asia [7,12] when compared to the outcome of the cases in the Americas, where systemic EBV-positive T-cell lymphoma of childhood was almost uniformly fatal.
Histology and Immunohistochemistry
Microscopically, systemic EBV-positive T-cell lymphoma of childhood can present as a diffuse infiltrate of small typicalappearing T-lymphocytes with cytotoxic characteristics within several end organs simultaneously. The most affected organs are the bone marrow, spleen, liver, and lymph nodes. There generally be an infiltrate of atypical T-lymphocytes in the bone marrow interstitium (Figure 1A), that are positive for CD3 by immunohistochemistry (Figure 1B) and EBER by in situ hybridization (Figure 1C). Significant hepatomegaly and splenomegaly may develop. A dense portal lymphohistiocytic infiltrate can be observed in the liver (Figure 1D), with a concurrent lymphohistiocytic infiltrate within the splenic sinusoids (Figure 1E). These neoplastic T-cells are also positive for CD3 (Figure 1F) and demonstrate diffuse EBER positivity (Figure 1G). Lymph nodes may also demonstrate dilation of their sinusoidal spaces with lymphocytes and hemophagocytic histiocytes (Figure 1H), and these lymphocytes and histiocytes are diffusely reactive for EBER (Figure 1I).
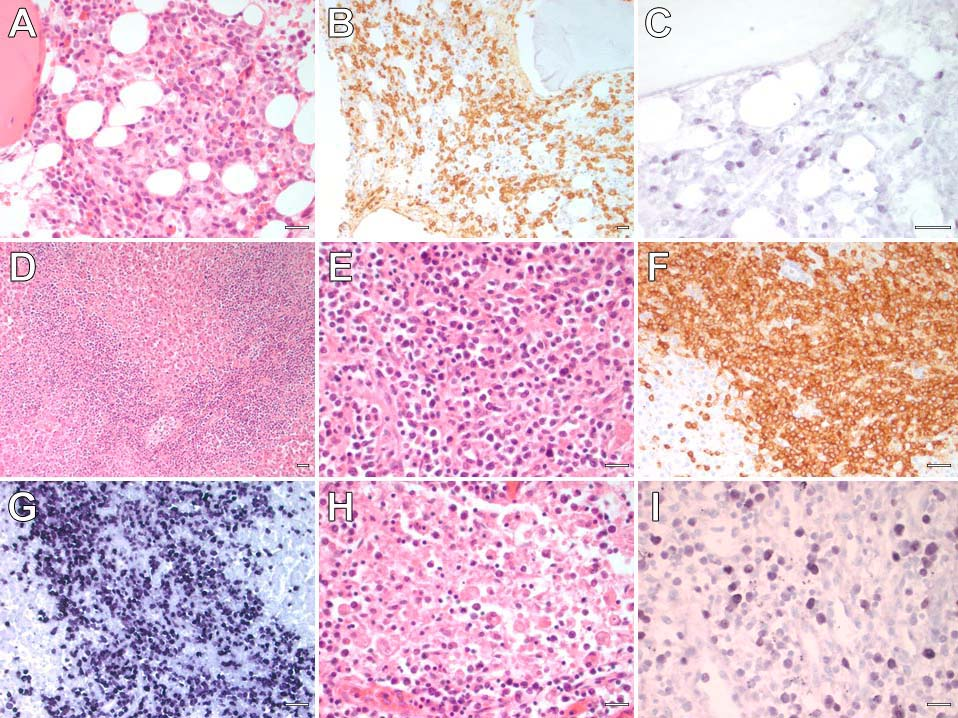
Figure 1. Hematoxylin and eosin (H/E) stained section of the bone marrow (A) showing an interstitial infiltrate of atypical T-lymphocytes highlighted by CD3 (B) and reactive for EBER (C). A section of the liver (H/E) shows a dense portal lymphoid infiltrate (D). Higher magnification shows that these atypical lymphocytes (E) are CD3-positive T-lymphocytes (F) with diffuse EBER reactivity (G). A dilated sinusoidal space in a lymph node (H) showing hemophagocytosis, and nuclear positivity for EBER (I). All scale bars = 25 micrometers.
Differential Diagnosis
There is extensive overlap in the clinical presentation between systemic EBV-positive T-cell lymphoma of childhood from EBVHLH [11,14,15]. Both demonstrate an infiltrate of monomorphic EBV-positive CD8(+) T-lymphocytes within multiple organs including the liver, spleen, lymph nodes and bone marrow [16]. However, in systemic EBV-positive T-cell lymphoma of childhood, the CD8(+) T-lymphocytes are neoplastic, showing monoclonal TCRβ and TCRγ rearrangements. Both entities may also show hemophagocytosis within these organs, suggestive of associated HLH. As this disease progresses, disseminated intravascular coagulopathy (DIC) may manifest in the terminal stages of multi-organ failure. Although DIC has been documented in the context of advanced HLH [16-19], its presence is not indicative of any distinction between systemic EBV-positive T-cell lymphoproliferative disorder of childhood and an associated EBV-positive hemophagocytic lymphohistiocytosis. There are several prognostic features are suggestive of a rapid and aggressive course, such as the presence of marked atypia of the neoplastic CD8(+) T-cells or the presence of circulating EBV DNA [9]. Ultimately, the terminal physiological events will manifest into multi-organ failure or HLH, or a combination of both [11,12,16].
Conclusions
EBV-positive T/NK-associated lymphoproliferative disorders of childhood are rare entities with distinctly different clinical outcomes. The most serious but rarest manifestation of primary T-cell EBV infection is systemic EBV-positive T-cell lymphoma of childhood, which appears to demonstrate a dichotomy in the outcome based on the geographical considerations [7,10,12]. Much of our knowledge about these rare EBV-associated diseases derives from case reports and case series detailing an aggressive clinical course with poor response to therapy resulting in death within days to weeks [20,21]. We hope this mini review can provide a more comprehensive overview of several possible manifestations of EBV-associated systemic T-cell lymphoproliferative disorders of childhood and add to the increasing body of evidence that extensive overlap occurs between the various entities of EBVassociated lymphoproliferative disorders.
References
2. Carbone A, Gloghini A, Dotti G. EBV-associated lymphoproliferative disorders: classification and treatment. The Oncologist. 2008 May;13(5):577-85.
3. Kimura H, Ito Y, Kawabe S, Gotoh K, Takahashi Y, Kojima S, et al. EBV-associated T/NK–cell lymphoproliferative diseases in nonimmunocompromised hosts: prospective analysis of 108 cases. Blood, The Journal of the American Society of Hematology. 2012 Jan 19;119(3):673-86.
4. Kawamoto K, Miyoshi H, Suzuki T, Kozai Y, Kato K, Miyahara M, et al. A distinct subtype of Epstein-Barr virus-positive T/NK-cell lymphoproliferative disorder: adult patients with chronic active Epstein-Barr virus infection-like features. Haematologica. 2018 Jun;103(6):1018-28.
5. Chiu TM, Lin YM, Wang SC, Tsai YG. Hypersensitivity to mosquito bites as the primary clinical manifestation of an Epstein–Barr virus infection. Journal of Microbiology, Immunology and Infection. 2016 Aug 1;49(4):613-6.
6. Cinbis M, Aysun S. Alice in Wonderland syndrome as an initial manifestation of Epstein-Barr virus infection. British Journal of Ophthalmology. 1992 May 1;76(5):316.
7. Cohen JI, Kimura H, Nakamura S, Ko YH, Jaffe ES. Epstein–Barr virusassociated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8–9 September 2008. Annals of Oncology. 2009 Sep 1;20(9):1472-82.
8. Ohga S, Ishimura M, Yoshimoto G, Miyamoto T, Takada H, Tanaka T, et al. Clonal origin of Epstein-Barr virus (EBV)-infected T/NKcell subpopulations in EBV-positive T/NK-cell lymphoproliferative disorders of childhood. Journal of Clinical Virology. 2011 May 1;51(1):31-7.
9. Hong M, Ko YH, Yoo KH, Koo HH, Kim SJ, Kim WS, et al. EBV-positive T/NK-cell lymphoproliferative disease of childhood. Korean Journal of Pathology. 2013 Apr;47(2):137.
10. Coffey AM, Lewis A, Marcogliese AN, Elghetany MT, Punia JN, Chang CC, et al. A clinicopathologic study of the spectrum of systemic forms of EBV-associated T-cell lymphoproliferative disorders of childhood: a single tertiary care pediatric institution experience in North America. Pediatric Blood & Cancer. 2019 Aug;66(8): e27798.
11. Chen G, Chen L, Qin X, Huang Z, Xie X, Li G, et al. Systemic Epstein-Barr virus positive T-cell lymphoproliferative disease of childhood with hemophagocytic syndrome. International Journal of Clinical and Experimental Pathology. 2014;7(10):7110-113.
12. Su IJ, Chen RL, Lin DT, Lin KS, Chen CC. Epstein-Barr virus (EBV) infects T lymphocytes in childhood EBV-associated hemophagocytic syndrome in Taiwan. The American Journal of Pathology. 1994 Jun;144(6):1219-1225.
13. Yoshii M, Ishida M, Hodohara K, Okuno H, Nakanishi R, Yoshida T, et al. Systemic Epstein-Barr virus-positive T-cell lymphoproliferative disease of childhood: Report of a case with review of the literature. Oncology Letters. 2012 Sep 1;4(3):381-4.
14. Smith MC, Cohen DN, Greig B, Yenamandra A, Vnencak-Jones C, Thompson MA, et al. The ambiguous boundary between EBV-related hemophagocytic lymphohistiocytosis and systemic EBV-driven T cell lymphoproliferative disorder. International Journal of Clinical and Experimental Pathology. 2014;7(9):5738-49.
15. Coffey AM, Lewis A, Marcogliese AN, Elghetany MT, Punia JN, Chang CC, et al. A clinicopathologic study of the spectrum of systemic forms of EBV-associated T-cell lymphoproliferative disorders of childhood: a single tertiary care pediatric institution experience in North America. Pediatric Blood & Cancer. 2019 Aug;66(8): e27798.
16. Keow JY, Stecho WM, Haig AR, Sangle NA. EBV-positive T/NKassociated lymphoproliferative disorders of childhood: A complete autopsy report. Indian Journal of Pathology and Microbiology. 2020 Jan 1;63(1):78-82.
17. Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang W, et al. Hemophagocytic lymphohistiocytosis: clinical analysis of 103 adult patients. Medicine. 2014 Mar;93(2):100-5.
18. Valade S, Azoulay E, Galicier L, Boutboul D, Zafrani L, Stepanian A, et al. Coagulation disorders and bleedings in critically ill patients with hemophagocytic lymphohistiocytosis. Medicine. 2015 Oct;94(40).
19. George MR. Hemophagocytic lymphohistiocytosis: review of etiologies and management. Journal of Blood Medicine. 2014;5: 69-86.
20. Hue SS, Oon ML, Wang S, Tan SY, Ng SB. Epstein–Barr virusassociated T-and NK-cell lymphoproliferative diseases: an update and diagnostic approach. Pathology. 2020 Jan 1;52(1):111-27.
21. Kim WY, Montes-Mojarro IA, Fend F, Quintanilla-Martinez L. Epstein-Barr virus-associated T and NK-cell lymphoproliferative diseases. Frontiers in Pediatrics. 2019 Mar 15;7:71.