Abstract
Amyotrophic Lateral Sclerosis (ALS) is a fatal progressive neurodegenerative disease characterized by the destruction of the motor neurons. It usually affects people between 40 and 60-year-old and the average survival from onset to death is 3–4 years. Despite the severity of the disease and the high health care and social costs, no cure or viable long-term effective treatment has been identified. Moreover, the failure to translate positive preclinical results from the SOD1 mouse model into clinical efficacy has raised questions about the translational suitability of this model. For this reason, the TDP-43 transgenic mouse model was created by overexpressing the mutant human TDP-43 gene, mutation directly related with ALS. In this study we characterized this mouse strain TDP-43, recognized as a preclinical mouse model for ALS disorder. We observed neuromuscular disorders, peripheral nerve electrophysiological impairment and histological anomalies at 3 months old. We also demonstrated that intrathecal injection of AAV1 expressing shRNA for casein kinase-1d (CK1d) attenuated the peripheral degenerative phenotype in this ALS model. Our data confirm that TDP-43 mouse strain is a robust and reproducible model to analyze the neuropathy disorders of ALS and that gene therapy silencing CK1d is a promising therapy for human ALS disorder, and can be used as a positive reference control for additional new drugs efficacy studies targeting ALS.
Introduction
Amyotrophic Lateral Sclerosis (ALS) is a fatal progressive neurodegenerative disease, which results in the destruction of upper and/or lower motor neurons in the brain and spinal cord. It usually affects people between 40 and 60-year-old and the average survival from onset to death is 3–4 years [1]. Despite the severity of the disease and the high health care and social costs, no cure or viable long-term effective treatment has been identified, with only two therapeutic agents (riluzole and edaravone) already approved by Food and Drug Administration but having limited efficacy or serving only for specific groups of patients [2].
In 2006, the trans-activating response region DNA binding protein of 43 kDa, known as TDP-43, was identified as one of the main hallmarks of sporadic and familial ALS, showing accumulations in the cytoplasm of cortical and motor neurons [3]. Mutations in this protein have been associated with cases of ALS, but also have been associated with tau protein-independent cases of frontotemporal dementia (FTD), generating the idea of a pathological spectrum between ALS and FTD based on alterations in TDP-43 and other related proteins [4]. Today, it is recognized that TDP-43 proteinopathy, characterized by hyperphosphorylation, truncation, ubiquitination, and/ or nuclear depletion in neurons, is the prominent and common pathological feature of sporadic and familiar ALS [1]. Furthermore, it is present in rare diseases such as Perry syndrome or Alexander disease, but also in the prevalent Alzheimer’s disease (AD) and recently in the limbic-predominant age-related TDP-43 encephalopathy [2,3]. In addition, TDP-43 pathology is a secondary feature of several other neurodegenerative disorders, including Parkinson’s disease, and Huntington’s disease, where its presence may aggravate the primary existing proteinopathy [4]. Increasing number of publications demonstrated that in ALS, TDP-43 concentration is formation [5,6]. Mitochondrial localization of TDP-43 depends on internal motifs M1 (aa 35–41), M3 (aa 146– 150), and M5 (aa 294–300), which consists of continuous increased in cytoplasm leading to cytoplasmic inclusion stretch of hydrophobic amino acids [7]. Owing to its poor in vitro solubility and high aggregation propensity, the complete structure of TDP-43 has remained elusive thus far. Several groups, however, have determined high resolution structures of some of its domains and its holistic structure is now evolving.
CK-1 was the first kinase identified to phosphorylate TDP-43 in vivo in more than 29 different sites, being 18 of them located in the C-terminal glycine-rich region [8]. Moreover, different stress signaling cause CK-1-dependent phosphorylation of TDP-43 triggering its cytosolic mislocalization and accumulation [9,10]. Furthermore, TDP-43 binds directly to and regulates the expression of CK-1ε mRNA [11].
Importantly, chemical inhibition of CK-1, mainly the δ and ε isoforms, has been proposed as potential treatment for different neurodegenerative diseases including ALS, FTD and Alzheimer’s disease [12]. In this way, and as showed in the Figure 1a, we developed an AAV1 expressing the shRNA for mouse CK-1δ in order to reduce the expression of CK- 1δ in motor neurons. We demonstrated that the silencing of CK-1δ by intrathecal injection significantly increased neuromuscular performances and reduced motor neuron degeneration attenuating ALS neuropathy. These changes were probably induced by the reduction of the TDP-43 phosphorylation and the accumulation and mislocalization of this protein. Our data confirm that the silencing of CK- 1δ can be used as a positive reference control for additional new drugs efficacy studies targeting ALS disorder.
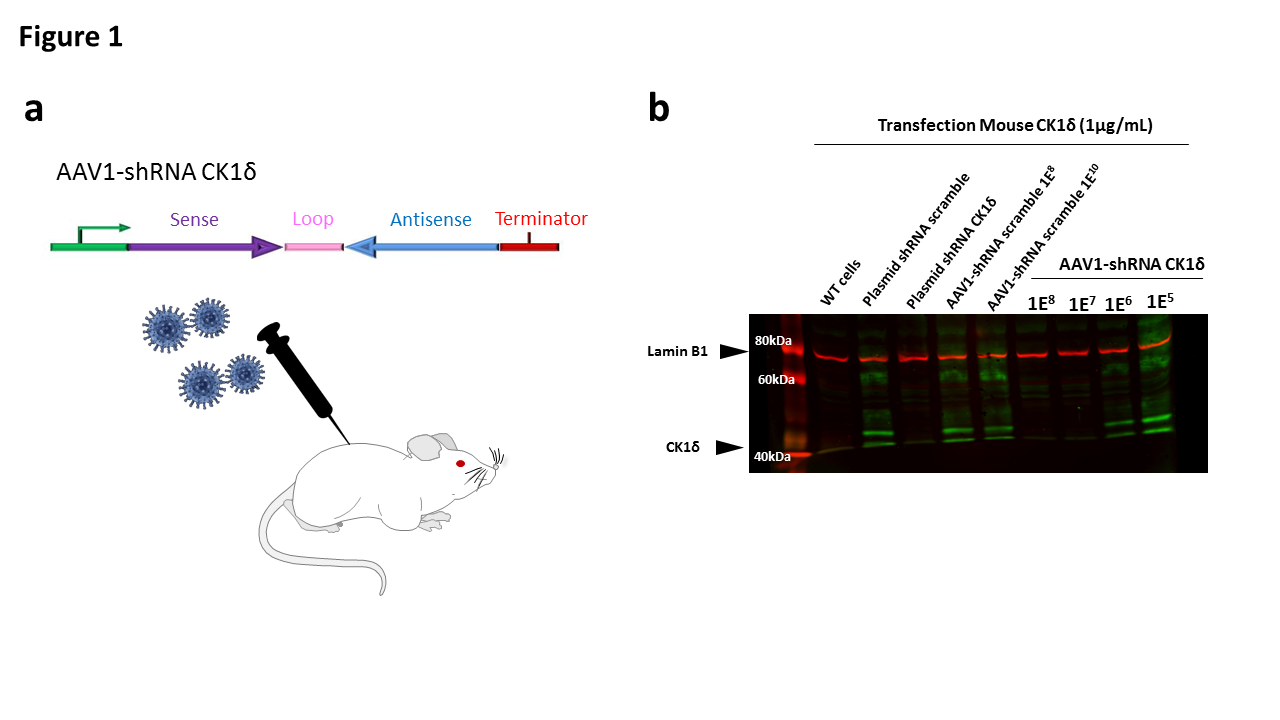
Figure 1. a. Schematic representation of the intrathecal viral particles injection. b. Western blot image of the in vitro validation of the plasmid and viral particles expressing shRNA – CK1δ
Materials and Methods
Cloning and virus production
The mouse CK-specific shRNA sequence TGATAAGTCGTATTGAGTA and shRNA scramble (control) sequences AGTTCCAGTACGGCTCCAA were separately cloned into a pAAV vector using XhoI and EcoRV restriction sites. Each cloning was validated by sequencing. Then, to produce high-titer adeno-associated virus (AAV1), three 15 cm dishes of 70-80% confluent HEK293T cells were transfected with 71 μg of pAAV expression vector, 20 μg of pAAV1 capsid and 40 μg of pHelper (Cell Biolabs, Inc.). 48 h later transfection, the medium was collected, pooled and centrifuged 15 min at 2000 rpm to spin down floating cells. In parallel, cells were scraped and collected in PBS. Then, cells were lysed using dry ice/ethanol bath and centrifuged 15 min at 5000 rpm to discard cell debris. The cleared supernatant and the cleared medium were pooled and filtrated using a 0.22 μm filter. The viral solution was filtrated through a cation-exchange membrane Mustang S acrodisc (Pall Corporation) to deplete empty particles and later, filtrated through an anion-exchange membrane Mustang Q acrodisc (Pall corporation) to retain AAV viral particles. Then, viruses were eluted and concentrated using centrifugal concentrators Amicon tube. The viral titer used was 1×1012 vg/mL.
Cell culture and transfection
Human embryonic kidney (HEK)-293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 2 mM L-glutamine, 100 U/ml penicillin/streptomycin, and 5% (v/v) heat inactivated Foetal Bovine Serum (FBS) (all from Invitrogen). Cells were maintained at 37°C in an atmosphere of 5% CO2, and were passaged every 3 or 4 days when they were 80-90% confluent. HEK-293T cells growing in 6-well dishes were transiently transfected with 1 μg of cDNA of mouse CK-1δ- flag using Lipofectamine method (Invitrogen). 48 hours after transfection, cells were infected with increasing concentration of viral particles. 24 hours after infection, cells were lysed and the expression level of CK-1δ was determined by western blot.
Western blot
Cell lysed samples were denaturalized and separated in 10% SDS-polyacrylamide gel and transferred onto nitrocellulose membranes. Membranes were blocked for 60 min with 5 mL of Licor Blocking Buffer. The following primary antibodies were incubated overnight at 4°C in the same blocking buffer: rabbit anti-lamin B1 (1:100, Sigma) and mouse anti-flag (1:100, Sigma). The following day the membranes were washed 3 times for 10 minutes in TBS Tween-20 (0.1% V/V) and the incubated for 1 hour at room temperature with the secondary fluorescence antibodies : donkey anti-rabbit IRDye 800 (1:10.000, Licor) and Goat anti-mouse IRDye 680 (1:10.000, Licor). After secondary antibody incubation, the membranes were washed three times for 10 min each with TBS Tween-20 (0.1% V/V). Visualization of the bands was performed using Licor scanning devise.
Animal housing
Male Prp-TDP43A315T mice and wild type control were kept in the A1 animal house facility. Mice were housed in ventilated and clear plastic boxes and subjected to standard light cycles (12 hours in 90-lux light, 12 hours in the dark). Baseline experiments started at 1 month old and ALS phenotype was characterized at 2 and 3 months old. All animal experiments were approved by the Comité d’Ethique en Expérimentation Animale du Languedoc- Roussillon, Montpellier, France, and the Ministère de l’Enseignement Supérieur et de la Recherche, Paris, France.
Intrathecal injection
In ketamine/xylazine anesthetized animals, a 10 μl Hamilton needle was gently and vertically (or tilt slightly 70-80°) inserted in the intersection of the L5- L6 intervertebral space. The angle was reduced to approximately 30° slowly when it connected the bone, then the needle was slipped into the intervertebral space. 8 μL of the AAV1-shRNA CK1d solution was injected at a speed of 1 μL/4 seconds. The needle was retained approximately 1 min after finishing AAV1 delivery.
Balance beam test
Balance beam test is a narrow “walking bridge” that rodents can cross to test balance and neurosensory coordination. The beam (thickness 6 cm) was elevated with the help of two feet with platforms to hold mice. The time required to cross the beam from side to side was quantified for each animal. Each animal underwent 3 trials/day at 5 minutes intervals. For each day, values from the 3 trials were averaged for each animal, normalized according to animal weight, and then averaged for each treated group.
Rotarod
A rotating rod apparatus was used to measure neuromuscular coordination and balance. Mice were first given a 2-day pretraining trial to familiarize them with the rotating rod. Latency to fall was measured at a successively increased speed from 4 to 40 rpm over a 300-second time period. Each animal underwent 3 trials a day. For each day, values from the 3 trials are averaged for each animal, normalized according to animal weight, and then averaged for each group.
Grip test
Neuromuscular strength of mice was assessed in standardized grip strength tests for hind limbs. Limbs grip strength was measured by supporting each rodent on a metal grid and pulling the animal’s tail toward a horizontal grid connected to a gauge. The maximum force (measured in newtons) exerted on the grid before the animal lost its grip was recorded, and the mean of 3 repeated measurements was calculated. All data were normalized according to animal weight.
Sciatic nerve electrophysiology
Standard electromyography was performed on mice anesthetized with ketamine/xylazine mixture. A pair of steel needle electrodes (AD Instruments. MLA1302) was placed subcutaneously along the nerve at the sciatic notch (proximal stimulation). A second pair of electrodes was placed along the tibial nerve above the ankle (distal stimulation). Supramaximal square-wave pulses, lasting 10 ms at 1 mA were delivered using a PowerLab 26T (AD Instruments). CMAP was recorded from the intrinsic foot muscles using steel electrodes. Both amplitudes and latencies of CMAP were determined. The distance between the 2 sites of stimulation was measured alongside the skin surface with fully extended legs, and NCVs were calculated automatically from sciatic nerve latency measurements. The left sciatic nerves were analyzed in this study.
Sciatic nerve histology by Transmission Electron Microscopy (TEM)
The sciatic nerves of all mice were fixed for 20 minutes in situ with 4 % PFA and 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3). Then, nerves were removed and post-fixed overnight in the same buffer. After washing for 30 minutes in 0.2 M PBS buffer, the nerves were incubated with 2% osmic acid in 0.1 M phosphate buffer for 90 minutes at room temperature. Then, samples were washed in 0.2 M PBS buffer, dehydrated using ethanol gradient solutions, and embedded in epoxy resin. Ultrathin (70-nm) cross sections were cut and stained with 1% uranyl acetate solution and lead citrate and analyzed using a HITACHI H7100 electron microscope at the COMET platform. The left sciatic nerves were analyzed in this study.
Statistics
Data are represented as mean ± SEM. Statistical significance was determined using a 2-tailed Student’s t test or 1-way ANOVA, followed by a Dunnett’s multiple comparison post-hoc test. A P value of less than 0.05 was considered significant. n= 5 animals/group.
Results
In vitro validation of shRNA for CK-1δ
In vitro HEK293T cells were transfected with 1 μg of cDNA mouse CK-1δ-flag. 24 hours after transfection, cells were infected with increasing concentrations of AAV1-shRNA CK-1δ or shRNA scramble (negative control). 48 hours after infection, cells were lysed and the concentration of mouse CK-1δ was determined by western blot using mouse anti-flag antibody. Lamin B1 was used as protein control loading.
Our data confirmed that the plasmid shRNA mouse CK- 1δ and the viral particles AAV1 expressing shRNA for CK- 1δ were able to decrease the expression of mouse CK-1δ in vitro (Figure 1b).
Decrease of neuromotor impairment in ALS mouse model induced by CK-1δ silencing
No mortality and no pathological clinical signs were observed during the study. As expected, significant decrease of body weight was observed in the TDP-43 groups compared to the control group at 3 months old. Moreover, a slight but significant difference of body weight was also observed between TDP-43 and TDP-43 + AAV shCK1d at 3 months old. No significant difference was observed in the TDP-43 + AAV shCK1d group compared to the control group (Figure 2a) suggesting an effect of the intrathecal AAV injection in the body weight loss induced by ALS phenotype.
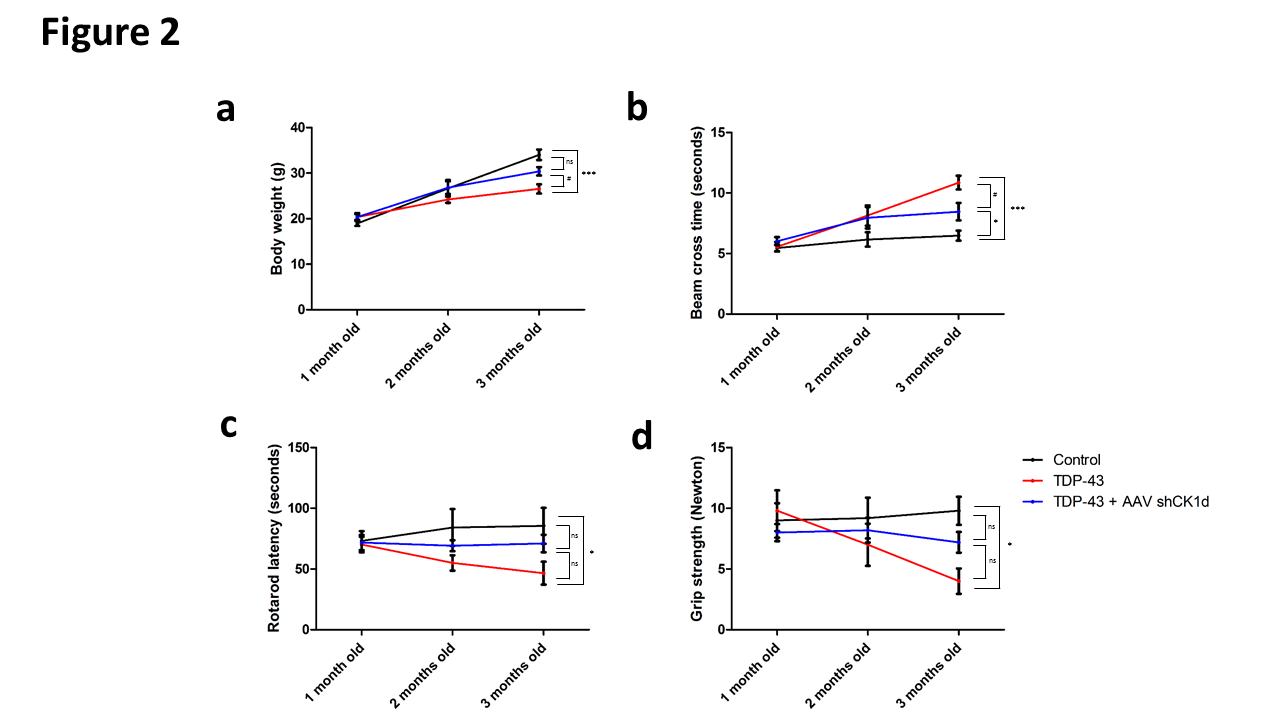
Figure 2. Analysis of the neuromuscular performance. a. Animal body weight of control, b. Balance beam cross time, c. Rotarod latency, and d. grip strength of control, TDP-43 and TDP-43 + AAV shRNA CK1δ groups at 1, 2 and 3 months old. Values are mean. Error bars indicate SEM. Statistical tests are repeated measures 1-way ANOVA test comparing groups to control (*) or TDP-43 (#) values. #* p<0.05, ##** p<0.01, *** p<0.001. ns= no significant. n=5 mice/group.
Walking performances were analyzed using balance beam test. Similar beam cross times were observed in control, TDP-43 and TDP-43 + shCK1δ groups at 1 month old (5.4 ± 0.3 s; 5.6 ± 0.4 s; 6.0 ± 0.3 s respectively). At 2 months old, the beam cross time was increased at 8.1 ± 0.9 seconds in the TDP-43 group mice whereas the cross time of control animals remained at 6.2 ± 0.6 s. A slight increase of the beam cross time was also observed in the TDP-43 + shCK1δ compared to control. However, no statistical differences were observed between all groups at this time point (Figure 2b). At 3 months old, the beam cross time of the TDP-43 group was significantly increased at 10.8 ± 0.6 seconds whereas the control animals presented a beam cross time of 6.5 ± 0.4 s. Moreover, the TDP-43 + shCK1δ group also presented a slight but significant increase of the cross time compared to the control animals at 3 months old. However, a statistical difference was observed between TDP-43 and TDP-43 + shCK1δ groups at 3 months old suggesting a partial protective effect of the viral particle injection in the walking impairment (Figure 2b).
Then, the neuromuscular strength was analyzed using grip test. Similar grip strengths were observed in control, TDP-43 and TDP-43 + shCK1δ groups at 1 month old (9.0 ± 1.4 N; 9.8 ± 1.7 N; 8.0 ± 0.7 N respectively). At 2 months old, the grip strength was decreased at 7.0 ± 1.7N in the TDP-43 group whereas the grip strength of control group remained at 9.2 ± 1.7N. However, no statistical difference was observed between TDP-43 and control groups at this time point. Moreover, no differences in the grip strength were observed in the TDP-43 + shCK1δ at 2 months old compared to the baseline (Figure 2c). At 3 months old, the grip strength of the TDP-43 group was significantly decreased at 4.0 ± 1.0 N whereas the control animals presented strength of 9.8 ± 1.2N. Moreover, even if the TDP-43 + shCK1δ group presented a slight decrease of the grip strength compared to the baseline, no statistical differences were observed between TDP-43 + shCK1δ versus control and TDP-43 animals at 3 months old (Figure 2c). Taken together these data also suggested a protective effect of the viral particle injection in the neuromuscular strength.
Finally, the coordination and balance were analyzed using rotarod. Similar rotarod latencies were observed in control, TDP-43 and TDP-43 + shCK1δ groups at 1 month old (73.2 ± 8.1 s; 70.0 ± 6.5 s; 71.6 ± 6.2 s respectively). At 2 months old, the rotarod latency was decreased at 55.0 ± 6.4s in the TDP-43 group whereas the latency of control group remained at 84.3 ± 15.0 N. However, no statistical difference was observed between TDP-43 and control groups at this time point. A slight increase of the rotarod latency was also observed in the TDP-43 + shCK1δ compared to control. However, no statistical differences were observed between all groups at 2 months (Figure 2d). At 3 months old, the rotarod latency of the TDP-43 group was significantly decreased at 47.0 ± 9.4 s whereas the control animals presented a latency of 85.7 ± 14.9s. Moreover, no statistical differences in the rotarod latency were observed in the TDP-43 + shCK1δ group compared to the control and TDP-43 animals at 3 months old (Figure 2d). Taken together these data also suggest a protective effect of the viral particle injection in the balance and motor coordination performance.
CK-1δ silencing increased the nerve conduction velocity in ALS
Compound muscle action potential amplitude and nerve conduction velocity were determined using standard electromyography in anesthetized mice to corroborate behavioral test data.
Concerning the CMAP amplitude, similar values were observed in control, TDP-43 and TDP-43 + shCK1δ groups at 1 months old (4.54 ± 0.52 mV; 4.93 ± 0.75 mV; 4.08 ± 0.66 mV respectively). At 2 and 3 months old, a slight but no significant decrease of the CMAP amplitude was observed in the TDP-43 compared to the control group. No differences were observed in the TDP43 + shCK1δ compared to the baseline and between all groups (Figure 3a).
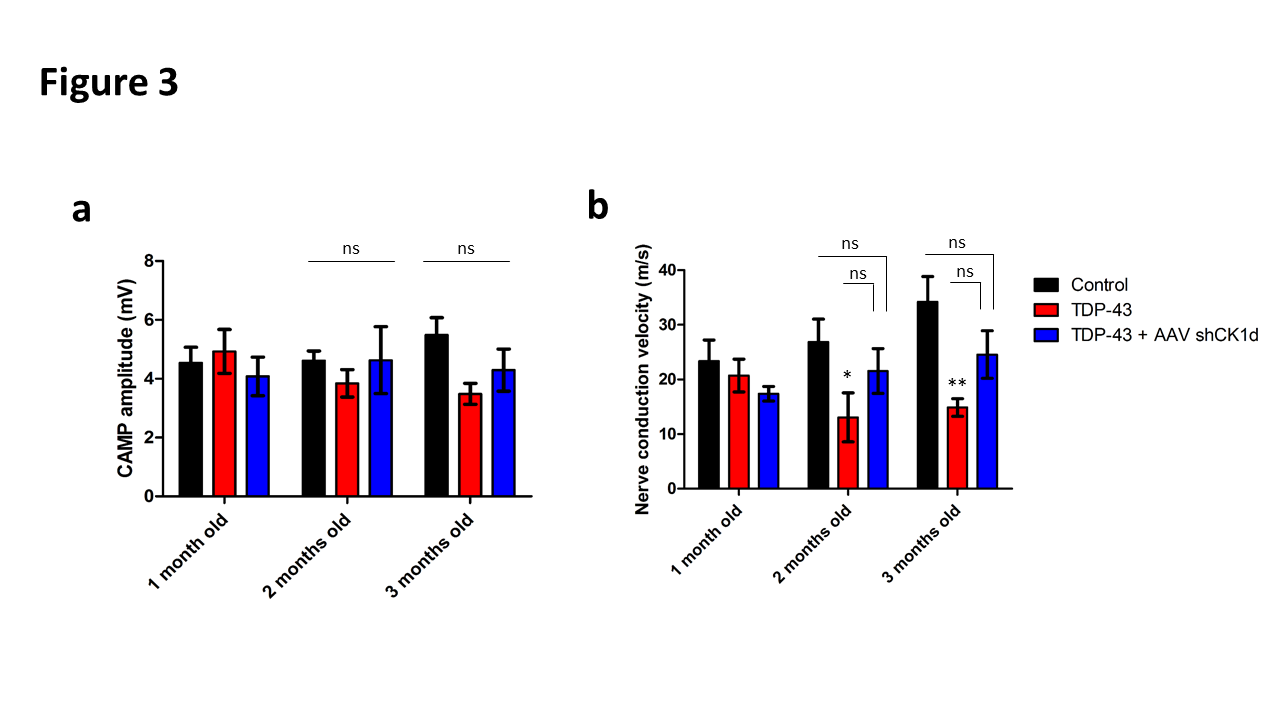
Figure 3. Analysis of the sciatic nerve conduction. a. compound action muscle potential (CAMP) analysis represented in mV and b. nerve conduction velocity represented in m/s of control, TDP-43 and TDP-43 + AAV shRNA CK1δ groups at 1, 2 and 3 months old. Values are mean. Error bars indicate SEM. Statistical tests are repeated measures 1-way ANOVA test comparing groups to control or TDP-43 values. * p<0.05, ** p<0.01, *** p<0.001. ns= no significant. n=5 mice/group.
Concerning the nerve conduction velocity (NCV), similar values were observed in control, TDP-43 and TDP-43 + shCK1δ groups at 1 months old (23.37 ± 3.85 m/s; 20.72 ± 3.01 m/s; 17.39 ± 1.33 m/s respectively). At 2 and 3 months old, the NCV was significantly decreased in the TDP-43 group, but not in the TDP-43 + shCK1δ group, compared to the control (Figure 3b). However, no statistical differences were observed between TDP-43 + shCK1δ and TDP- 43 groups at these time points suggesting that the CK1δ silencing attenuated, but not stopped, the peripheral nerve degenerative phenotype of ALS.
CK-1δ silencing attenuated axonal loss of peripheral nerves
Finally, histological analysis of sciatic nerve was performed to characterize the morphology and number of motor neurons of TDP-43 mouse model and to determine the efficacy of AAV1-shRNA CK1δ in the ALS peripheral neuropathy.
Significant reduction of the axonal diameter was observed in the TDP-43 group compared to control group at 3 months old. Moreover, a slight but significant reduction of the axonal diameter was also observed in the TDP4-3- CK1δ compared to the control, but this reduction was statistically different compared to the TDP-43 group (Figure 4a and 4b). Also significant reduction of the number of axons was observed in the TDP-43 group mice compared to control mice at 3 months old. Moreover, a slight but significant reduction of the number of axons was also observed in the TDP4-3- CK1δ compared to the control, but this reduction was statistically different compared to the TDP-43 group (Figure 4c)
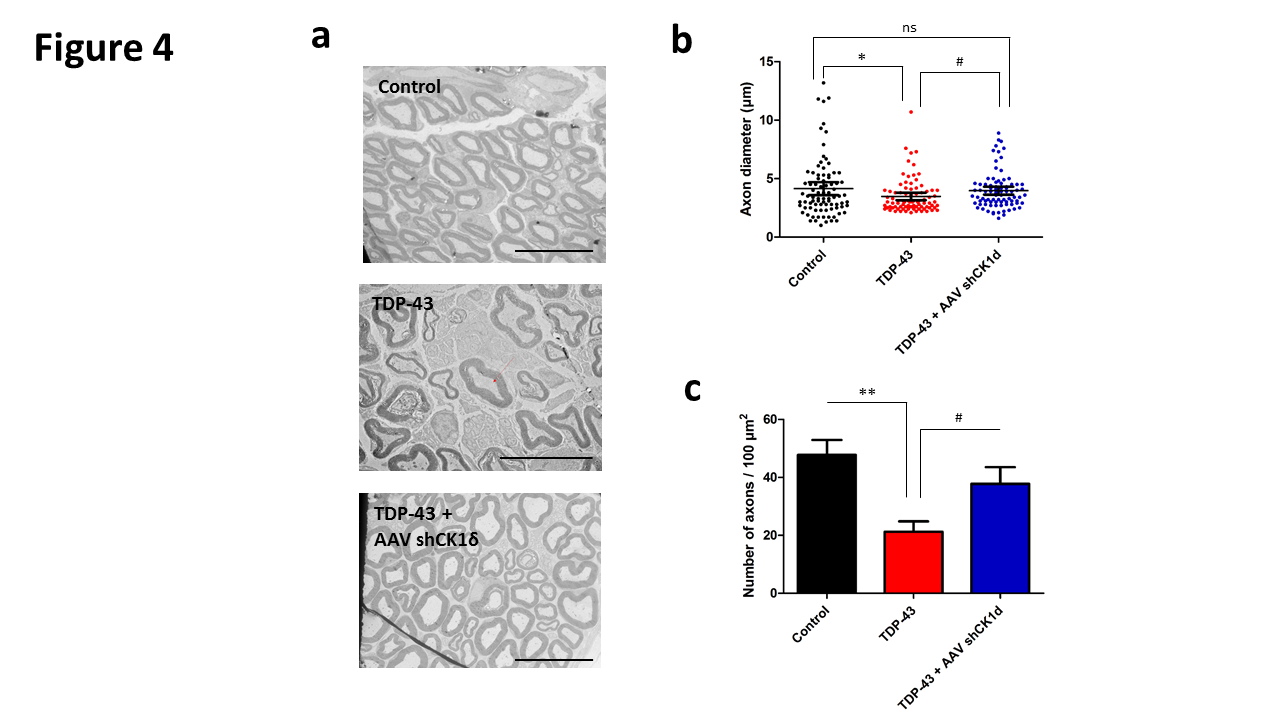
Figure 4. Histological analysis of sciatic nerve. a. Representative image of transmission electron microscopy of sciatic nerve of control, TDP-43 and TDP-43 + AAV shRNA CK1δ mice at 3 months old. Scale bar: 10 µm. b. Analysis of the axon diameter and, b. quantification of the number of axons par 100 µm2 of control, TDP-43 and TDP-43 + AAV shRNA CK1δ groups at 3 months old. Values are mean. Error bars indicate SEM. Statistical tests are repeated measures 1-way ANOVA test comparing groups to control or TDP-43 values. # * p<0.05, ** p<0.01. ns= no significant. n=5 mice/group.
Taken together, the histological analysis corroborated the behavioral and electrophysiology data confirming that the silencing of CK-1δ attenuated, but not stopped, the peripheral nervous system degeneration widely described in ALS disorder.
Discussion
The development of treatments for ALS has been hindered by a shortage of preclinical tools (animal models, cellular assays, biomarkers, etc.). The first mouse model of the disease was developed in 1994 by Gurney and collaborators [14] following the identification of mutations in the copper/zinc superoxide dismutase 1 (SOD1) gene linked to 20% of familial ALS cases [13]. This widely used mouse model, genetically engineered to overexpress a mutant form of the human SOD1 gene harboring the ALSassociated glycine to alanine mutation at amino acid 93 (SOD1 G93A), recapitulated many of the pathological features of ALS in humans and it promised to become an important in vivo screening tool for ALS therapies. Unfortunately, with the exception of riluzole [14], no effective therapy for ALS patients has been identified to date in the SOD1 mouse [15]. The failure to translate positive preclinical results from the SOD1 mouse model into clinical efficacy has raised questions about the translational suitability of this model. For this reason, the first TDP-43 transgenic mouse model was developed by overexpressing the mutant human TDP-43 gene, harboring the alanine to threonine mutation at amino acid 315, under the control of the mouse prion promoter (Prp-TDP43A315T) [16]. Prp-TDP43A315T mice reportedly developed a progressive and fatal neurodegenerative disease with pathology reminiscent of ALS. Additionally, several important features of the human disease were apparently replicated in these mice. These included gait abnormalities, selective vulnerability of cortical and spinal motor neurons, and ubiquitin aggregate pathology albeit without the presence of cytoplasmic TDP-43 aggregates [16] that occurs in humans.
Krach and collaborators described in 2018 a new family of potent CK-1δ and dual CK-1δ/ε inhibitors with high selectivity score over other kinases based on the modification of the benzothiazole scaffold [11]. They were able to decrease the TDP-43 phosphorylation both in cellular models and in vivo using a Drosophila transgenic model [17]. Moreover, two of these candidates, named as IGS-2.7 and IGS-2.37, also showed a decrease on TDP- 43 phosphorylation and nuclear localization using a cellbased model of human lymphoblast from FTD patients carrying a progranulin (GRN) mutation [18]. Therefore, Martínez-González and collaborators published in 2020 that these inhibitors of CK-1δ were able to modulate TDP- 43 proteinopathy in vivo, becoming a good therapeutic strategy for the severe ALS. They demonstrated that the treatment of the ALS preclinical model TDP-43 with a CK-1δ inhibitor decreased of TDP-43 phosphorylation, inducing motor neuron survival and decreasing astroglial and microglial reactivity [19]. This publication served as a solid proof of concept for the potential therapy of ALS with CK-1δ or dual CK-1δ/ε inhibitors showing a promising neuroprotective effect [19,20]. For this reason, in this study we developed an AAV1 expressing the shRNA for mouse CK-1δ in order to reduce the expression of CK-1δ in motor neurons looking to reproduce Martinez-Gonzalez results from a gene therapy point of view. We demonstrated that the silencing of CK-1δ by intrathecal injection reduced the motor neuron degeneration attenuating ALS neuropathy and corroborating Martinez-Gonzalez previous results. Our data demonstrated that the knockdown of CK-1δ using in vivo shRNA was able to partially reduce, but not to stop, the ALS phenotype in the TDP-43 mouse model. For this reason, we concluded that the silencing of CK-1δ is not perfect solution to ALS but a good attempt fighting against ALS in mouse models.
The study of the post-translational regulation of TDP-43 has placed the phosphorylation of this protein at specific residues, which is dependent on certain protein kinases, as a key event for regulating its cellular activities and also for its dysregulation associated with pathological conditions [21,22]. In fact, phospho-TDP-43 is distributed in brain areas of FTD and ALS patients [21]. Different kinases have been recently involved in TDP-43 phosphorylation. Among these, protein casein kinase-1 [22], tau tubulin kinase 1 [23] and cell division cycle kinase 7 [22] are the best characterized.
As published by Gou and collaborators in 2020 [24], aberrant phosphorylation of various ALS-related proteins by kinases could affect the localization and function of these proteins. However, the exact role these alterations play in motor neuron degeneration remains elusive. Confirming our hypothesis, multiple studies showed that mutations in genes encoding different kinases can cause or confer susceptibility to ALS, suggesting that alterations in the function of specific kinases and/or their downstream targets are vital to motor neuron survival. Taken together, these data suggest a potential key role of kinases in ALS genetics and pathophysiology [24].
In motor neurons, TDP-43 has also been shown in humans to be a low molecular weight neurofilament mRNAbinding protein [25]. It has also shown to be a neuronal activity response factor in the dendrites of hippocampal neurons suggesting possible roles in regulating mRNA stability, transport and local translation in neurons [26]. The TDP-43 protein contains 414 amino acids and the encoding gene TARDBP is located on the chromosome number 1. It comprises of an N-terminal region (aa 1–102) with a nuclear localization signal (NLS, aa 82–98), two RNA recognition motifs: RRM1 (aa 104–176) and RRM2 (aa 192–262), a nuclear export signal (NES, aa 239–250), a C-terminal region (aa 274–414) which encompasses a prion-like glutamine/asparagine-rich (Q/N) domain (aa 345–366) and a glycine-rich region (aa 366–414) [26,27]. TDP-43 is predominantly localized in the nucleus but also shuttles to the cytoplasm for some of its functions [28]. In ALS, there is an increase in the cytoplasmic TDP-43 concentration leading to cytoplasmic inclusion formation [29,30].
Conclusion
In this study we described the neuromotor disorders, peripheral nervous system electrophysiological impairment and histological anomalies observed in the preclinical ALS mouse model TDP-43 at 3 months old. Moreover, we demonstrated that intrathecal injection of AAV1 expressing shRNA for CK-1δ attenuated the peripheral nerve degeneration of ALS model. Our data confirm that TDP-43 mouse strain is a robust and reproducible model to analyze the neuropathy disorders of ALS and that gene therapy silencing CK1δ is a promising therapy for human ALS disorder. This silencing strategy can be used as a positive reference control for additional new drugs efficacy studies targeting ALS.
References
2. Palomo V, Tosat-Bitrian C, Nozal V, Nagaraj S, Martin- Requero A, Martinez A. TDP-43: a key therapeutic target beyond amyotrophic lateral sclerosis. ACS Chemical Neuroscience. 2019 Feb 20;10(3):1183-96.
3. Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant agerelated TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019 Jun 1;142(6):1503-27.
4. Davis SA, Gan KA, Dowell JA, Cairns NJ, Gitcho MA. TDP-43 expression influences amyloidß plaque deposition and tau aggregation. Neurobiology of Disease. 2017 Jul 1;103:154-62.
5. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006 Oct 6;314(5796):130-3.
6. Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. Journal of Biological Chemistry. 2008 May 9;283(19):13302-9.
7. Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nature Medicine. 2016 Aug;22(8):869-78.
8. Liachko NF, McMillan PJ, Guthrie CR, Bird TD, Leverenz JB, Kraemer BC. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Annals of Neurology. 2013 Jul;74(1):39-52.
9. Nonaka T, Suzuki G, Tanaka Y, Kametani F, Hirai S, Okado H, Miyashita T, Saitoe M, Akiyama H, Masai H, Hasegawa M. Phosphorylation of TAR DNA-binding protein of 43 kDa (TDP-43) by truncated casein kinase 1d triggers mislocalization and accumulation of TDP-43. Journal of Biological Chemistry. 2016 Mar 11;291(11):5473- 83.
10. Hicks DA, Cross LL, Williamson R, Rattray M. Endoplasmic reticulum stress signalling induces casein kinase 1-dependent formation of cytosolic TDP-43 inclusions in motor neuron-like cells. Neurochemical Research. 2019 Jul 6:1-1.
11. Krach F, Batra R, Wheeler EC, Vu AQ, Wang R, Hutt K, Rabin SJ, Baughn MW, Libby RT, Diaz-Garcia S, Stauffer J. Transcriptome–pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathologica. 2018 Sep;136(3):405-23.
12. Gu J, Hu W, Tan X, Qu S, Chu D, Gong CX, Iqbal K, Liu F. Elevation of casein kinase 1e associated with TDP- 43 and tau pathologies in Alzheimer’s disease. Brain Pathology. 2020 Mar;30(2):283-97.
13. Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993 Mar;362(6415):59-62.
14. Gurney ME, Cutting FB, Zhai P, Andrus PK, Hall ED. Pathogenic mechanisms in familial amyotrophic lateral sclerosis due to mutation of Cu, Zn superoxide dismutase. Pathologie-biologie. 1996 Jan 1;44(1):51-6.
15. Gordon PH, Meininger V. How can we improve clinical trials in amyotrophic lateral sclerosis?. Nature Reviews Neurology. 2011 Nov;7(11):650-4.
16. Wegorzewska I, Bell S, Cairns NJ, Miller TM, Baloh RH. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proceedings of the National Academy of Sciences. 2009 Nov 3;106(44):18809-14.
17. Cozza G, Pinna LA. Casein kinases as potential therapeutic targets. Expert opinion on therapeutic targets. 2016 Mar 3;20(3):319-40.
18. Gu J, Hu W, Tan X, Qu S, Chu D, Gong CX, et al. Elevation of casein kinase 1e associated with TDP-43 and tau pathologies in Alzheimer’s disease. Brain Pathology. 2020 Mar;30(2):283-97.
19. Martínez-González L, Rodríguez-Cueto C, Cabezudo D, Bartolomé F, Andrés-Benito P, Ferrer I, et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1d kinase inhibitor treatment. Scientific Reports. 2020 Mar 10;10(1):1-2.
20. Salado IG, Redondo M, Bello ML, Perez C, Liachko NF, Kraemer BC, et al. Protein kinase CK-1 inhibitors as new potential drugs for amyotrophic lateral sclerosis. Journal of Medicinal Chemistry. 2014 Mar 27;57(6):2755- 72.
21. Guedes ÁC, Santin R, Costa AS, Reiter KC, Hilbig A, Fernandez LL. Distinct Phospho-TDP-43 brain distribution in two cases of FTD, one associated with ALS. Dementia & Neuropsychologia. 2017 Sep;11(3):249-54.
22. Liachko NF, McMillan PJ, Guthrie CR, Bird TD, Leverenz JB, Kraemer BC. CDC7 inhibition blocks pathological TDP-43 phosphorylation and neurodegeneration. Annals of Neurology. 2013 Jul;74(1):39-52.
23. Liachko NF, McMillan PJ, Strovas TJ, Loomis E, Greenup L, Murrell JR, et al. The tau tubulin kinases TTBK1/2 promote accumulation of pathological TDP-43. PLoS Genetics. 2014 Dec 4;10(12):e1004803.
24. Guo W, Vandoorne T, Steyaert J, Staats KA, Van Den Bosch L. The multifaceted role of kinases in amyotrophic lateral sclerosis: genetic, pathological and therapeutic implications. Brain. 2020 Jun 1;143(6):1651-73.
25. Wang IF, Wu LS, Chang HY, Shen CK. TDP-43, the signature protein of FTLD-U, is a neuronal activityresponsive factor. Journal of Neurochemistry. 2008 May;105(3):797-806.
26. Cohen TJ, Lee VM, Trojanowski JQ. TDP-43 functions and pathogenic mechanisms implicated in TDP- 43 proteinopathies. Trends in Molecular Medicine. 2011 Nov 1;17(11):659-67.
27. Jiang LL, Zhao J, Yin XF, He WT, Yang H, Che MX, et al. Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Scientific Reports. 2016 Mar 31;6(1):1-1.
28. Ayala YM, Zago P, D’Ambrogio A, Xu YF, Petrucelli L, Buratti E, et al. Structural determinants of the cellular localization and shuttling of TDP-43. Journal of Cell Science. 2008 Nov 15;121(22):3778-85.
29. Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006 Oct 6;314(5796):130-133.
30. Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. Journal of Biological Chemistry. 2008 May 9;283(19):13302-9.