Abstract
Schwann cells produce myelin sheath around peripheral nerve axons. Myelination is critical for rapid propagation of action potentials, as illustrated by the large number of acquired and hereditary peripheral neuropathies, such as diabetic neuropathy or Charcot-Marie-Tooth diseases, that are commonly associated with a process of demyelination. Peripheral neuropathy is a major complication of diabetes, and the pathomechanism of the disease remains poorly studied. Here, we studied the progressive peripheral neurodegeneration characterized by a demyelinating process and hearing impairment observed in the diabetic mouse model db/db and we determined the efficacy of daily resveratrol (RSV) treatment in the neuromotor impairment and hearing loss. Moreover, we previously validated the efficacy of resveratrol treatment in a fast screening model of peripheral neuropathy, the sciatic nerve crush model. Our results confirm that these mice represent a robust and validated model to study the peripheral neuropathy induced by diabetes and open a potential opportunity to develop new pharmacological candidates targeting diabetic neuropathy.
Keywords
Diabetic, Diabetic neuropathy, Nerve injury
Introduction
In the peripheral nervous system (PNS), Schwann cells (SCs) are responsible for myelin production, which contributes to axonal protection and allows for efficient action potential transmission [1,2]. Unfortunately, acquired and hereditary demyelinating diseases of the PNS are numerous and affect an increasing number of people [3]. The etiologies of acquired and hereditary peripheral nerve diseases are diverse, but they all result in demyelination and subsequent neuronal death. Thus, an important challenge is to understand the cellular and molecular events that underlie demyelination of SCs.
Acquired demyelinating diseases are the most common, as they include diabetic peripheral neuropathy [4,5], drug-related peripheral neuropathies, leprosy, and inflammatory neuropathies [6]. Diabetic peripheral neuropathy is a major complication of diabetes type 1 and 2 and a cause of considerable morbidity [7,8]. Indeed, it has been reported that at least 50% of diabetic patients develop one or several forms of diabetic neuropathy within 25 years after diagnosis [7]. This neuropathy affects both myelinated SCs and peripheral axons/neurons, leading to changes in nerve conduction, and it is often associated with demyelination in the long term [9]. However, the physiological and molecular mechanisms that lead to these nerve defects remain unclear.
Diabetic neuropathy refers to various types of nerve damage associated with diabetes mellitus. Symptoms depend on the site of nerve damage and can include motor changes such as weakness; sensory symptoms such as numbness, tingling, or pain; or autonomic changes such as urinary symptoms. These changes are thought to result from microvascular injury involving small blood vessels that supply nerves. Relatively common conditions which may be associated with diabetic neuropathy include distal symmetric polyneuropathy, third, fourth, or sixth cranial nerve palsy, mononeuropathy, mononeuropathy multiplex, diabetic amyotrophy, and autonomic neuropathy.
High oxidative stress has been universally observed during diabetes mellitus disorder and peripheral diabetic neuropathy has also been directly associated to increase of ROS. For this reason, we hypothesized that resveratrol (RSV), a potent antioxidant able to scavenge a big number of oxygen species [10,11], could be an effective therapeutic agent to block the activation of diabetic neurodegenerative processes of peripheral nervous system. In this way, we studied the progressive neuromotor impairment and hearing loss of the diabetic mouse model db/db using behavioral tests, electrophysiological recording and transmission electron microscopy histology of sciatic nerve in control, diabetic+vehicle and diabetic+RSV treated animals. Our results suggest that db/db mouse reproduce the features of the neurodegenerative process that occurs in the human diabetic and that antioxidant treatment reduces the neuropathological phenotype. Moreover, these results were also confirmed using a fast screening model of nerve injury by crush, suggesting that ROS play a key role in Schwann cell demyelination and axonal degeneration during peripheral neuropathies. Therefore, db/db mouse seems an appropriate model to study the underlying mechanisms of diabetes-induced peripheral nervous system neurodegeneration and to develop new pharmacological candidates targeting diabetic neuropathy.
Materials and Methods
Animal housing and drug administration
Male C57BL/6J mice for nerve injury model, male db/ db diabetic mice [homozygous BKS(D)- Leprdb+/+/ JOrlRj, Janvier Labs] and male non-diabetic group control [heterozygous BKS(D)- Leprdb+/–/JOrlRj, Janvier Labs] were kept in the A1 animal house facility. Mice were housed in ventilated and clear plastic boxes and subjected to standard light cycles (12 hours in 90- lux light, 12 hours in the dark). In induced nerve injury model, experiments started at 2 months old. In diabetic mice, experiments started at 4 months of age (baseline) and finished at 6 months old.
In diabetic mice, Resveratrol (Sigma Aldrich, R5010- 100MG) was diluted in the drinking water at 100 μg/mL, and new solution was prepared every week during 8 weeks of treatment. The diabetic animals were treated with RSV from 4 months old to 6 months old. In nerve injury model, RSV was subcutaneously injected at 10 mg/kg, diluted in PBS + 2% DMSO, once a day during 4 days. All animal experiments were approved by the Comité d’Ethique en Expérimentation Animale du Languedoc-Roussillon, Montpellier, France, and the Ministère de l’Enseignement Supérieur et de la Recherche, Paris, France.
Sciatic nerve injury
Two months old mice were anesthetized with ketamine/ xylazine mixture and placed under a Stemi2000 microscope (Zeiss). Incision area was shaved and cleaned using betadine solution. After incision, the gluteus superficialis and biceps femoris muscles were separated to reveal the cavity traversed by the sciatic nerve. The nerve was crushed three times using forceps to induce sciatic nerve degeneration and neuropathy. After crush, the nerve was replaced into the cavity, the muscles were readjusted, and the wound was closed using clips.
Balance beam test
Balance beam test is a narrow “walking bridge” that rodents can cross to test balance and neurosensory coordination. The beam (thickness 6 cm) is elevated with the help of two feet with platforms to hold mice. The time required to cross the beam from side to side is quantified. Each animal underwent 3 trials a day at 5 minutes intervals. For each day, values from the 3 trials were averaged for each animal, normalized according to animal weight, and then averaged for each treated group.
Rotarod
A rotating rod apparatus was used to measure neuromuscular coordination and balance. Mice were first given a 2-day pretraining trial to familiarize them with the rotating rod. Latency to fall was measured at a successively increased speed from 4 to 40 rpm over a 300- second time period. Each animal underwent 3 trials a day. For each day, values from the 3 trials are averaged for each animal, normalized according to animal weight, and then averaged for each group.
Grip test
Neuromuscular strength of mice was assessed in standardized grip strength tests for hind limbs. Limbs grip strength was measured by supporting each rodent on a metal grid and pulling the animal’s tail toward a horizontal grid connected to a gauge. The maximum force (measured in newtons) exerted on the grid before the animal lost its grip was recorded, and the mean of 3 repeated measurements was calculated. All data were normalized according to animal weight.
Sciatic nerve electrophysiology
Standard electromyography was performed on mice anesthetized with ketamine/xylazine mixture. A pair of steel needle electrodes (AD Instruments. MLA1302) was placed subcutaneously along the nerve at the sciatic notch (proximal stimulation). A second pair of electrodes was placed along the tibial nerve above the ankle (distal stimulation). Supramaximal square-wave pulses, lasting 10 ms at 1 mA for mice were delivered using a PowerLab 26T (AD Instruments). CMAP was recorded from the intrinsic foot muscles using steel electrodes. Both amplitudes and latencies of CMAP were determined. The distance between the 2 sites of stimulation was measured alongside the skin surface with fully extended legs, and NCVs were calculated automatically from sciatic nerve latency measurements. The left sciatic nerves were analyzed in this study.
Sciatic nerve histology by Transmission Electron Microscopy (TEM)
The sciatic nerves of all mice were fixed for 20 minutes in situ with 4% PFA and 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.3). Then, nerves were removed and post- fixed overnight in the same buffer. After washing for 30 minutes in 0.2 M PBS buffer, the nerves were incubated with 2% osmic acid in 0.1 M phosphate buffer for 90 minutes at room temperature. Then, samples were washed in 0.2 M PBS buffer, dehydrated using ethanol gradient solutions, and embedded in epoxy resin. Ultrathin (70-nm) cross sections were cut and stained with 1% uranyl acetate solution and lead citrate and analyzed using a HITACHI H7100 electron microscope at the COMET platform. The left sciatic nerves were analyzed in this study.
Auditory brainstem responses (ABR)
For ABR studies, mice were anesthetized using ketamine/ xylazine mixture and body temperature is regulated using a heatingpad at 37°C. Then, earphones were placed in the left ear of each mouse, an active electrode was placed in the vertex of the skull, a reference electrode under the skin of the mastoid bone and a ground electrode was placed in the neck skin. The stimuli consisted of tone pips of five frequencies (4 kHz, 8 kHz, 16 kHz, 24 kHz and 32 kHz) at various sound levels (from 0 to 90 dB) ranging to cover the mouse auditory frequency range. ABR measures of each animal were performed individually and using OtoPhyLab system. Evoked potentials were extracted by the signal averaging technique for each noise level and ABR thresholds for each frequency were determined using OtoPhyLab software. The left ears were analyzed in this study.
Plasma TNF-α and IL-6 quantification
In nerve crush study, each animal was anesthetized using ketamine/xylazine, 2 mL of blood was sampled by cardiac puncture and collected in a tube containing EDTA as anticoagulant. Samples were centrifuge for 15 minutes at 1000 × g (or 3000 rpm) at 2-8°C within 30 minutes of collection. The supernatant (plasma) was stored at -20°C. Plasma was diluted at 1/10 and TNF-α and IL-6 quantification for each animal was performed in duplicated by ELISA method.
Statistics
Data are represented as mean ± SEM. Statistical significance was determined using a 2-tailed Student’s t test or 1-way ANOVA, followed by a Dunnett’s multiple comparison post-hoc test. A P value of less than 0.05 was considered significant. n=4 animals/group for nerve crush study and n=6 animals/group for diabetic study.
Results
RSV treatment attenuated neuromotor and electrophysiological impairment after nerve injury
The neuromuscular performance and strength were determined using two different behavioral tests.
Using grip test, similar neuromuscular strength was observed in control, nerve injury +vehicle and nerve injury +RSV groups at baseline (D1 – before crush). However, a significant decrease of the grip strength at 2.8 ± 0.7 N was observed in the nerve injury+vehicle group mice whereas the grip strength of control animals remained at 7.5 ± 1.4N. A slight and no significant decrease was also observed in the nerve injury+RSV compared to control and vehicle groups (Figure 1A).
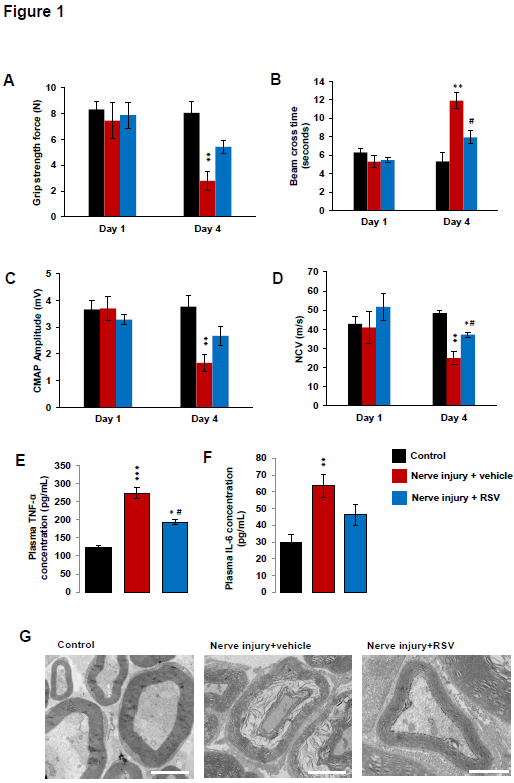
Figure 1. Peripheral neuropathy observed in nerve injury mice. (A) Grip strength, (B) Balance beam cross time, (C) sciatic nerve compound muscle action potential (CMAP) amplitude, (D) sciatic nerve conduction velocity, (E) plasma TNF-α concentration and (F) plasma IL-6 concentration of control (black bars), nerve injury+vehicle mice (red bars) and nerve injury+RSV (blue bars) at the day 1 (baseline, before nerve crush) and three days after nerve injury (day 4). (G) TEM sciatic nerve imaging of control, nerve injury+vehicle and nerve injury+RSV. Scale bar: 5 μm. Error bars indicate SEM. Statistical tests are repeated measures two-way ANOVA test comparing groups to control (*) or vehicle (#) values. #* p<0.05, ** p<0.01, *** p<0.001. n=4
The walking performance and equilibrium were then evaluated using balance beam test. Similar balance beam cross times were observed in control, nerve injury+vehicle and nerve injury+RSV groups at the baseline (D1 – before nerve crush). However, the beam cross time was significantly increased at 11 ± 2 seconds in the nerve injury+vehicle group mice whereas the cross time of control animals remained at 5 ± 1 seconds. Concerning the nerve injury+RSV group, the beam cross times was also increased at 8 ± 1 seconds but this increase was not significant compared to the control group. Interestingly, a statistical difference was observed between vehicle and RSV group at day 4 (Figure 1B).
Sciatic nerve conduction anomalies were observed three days after nerve injury. Similar compound muscle action potential (CMAP) amplitudes of sciatic nerve were observed in control, nerve injury+vehicle and nerve injury+RSV groups (around 3.6 mV) at baseline (D1 – before nerve injury). However, a significant decrease of the action potential amplitude at 1.7 ± 0.3 mV was observed in the nerve injury group mice whereas the amplitude of control animals remained at 3.7 ± 0.4 mV. A decrease of the CMAP amplitude was also observed in the nerve injury+RSV group but this decrease was no significant compared to the control and vehicle groups (Figure 1C).
Concerning the nerve conduction velocity (NCV), similar velocities of sciatic nerve were observed in control, nerve injury+vehicle and nerve injury+RSV groups at baseline (D1). However, a decrease of the nerve conduction velocity at 24.9 ± 3.5 m/s was observed in the nerve injury group mice whereas the velocity of control animals remained at 41.1 ± 8.6m/s. Moreover, a significant decrease of the NCV of the RSV group was also observed at day 4 compared to the control group. A statistical difference was observed between RSV and vehicle group suggesting a protective effect of RSV in the neuromotor impairment induced by nerve injury (Figure 1D).
RSV treatment reduced plasma inflammatory biomarkers and Schwann cell demyelination
ELISA analysis of plasma demonstrated a significant increase of the inflammatory biomarker TNF-α at 223.7 ± 15.3 pg/mL of plasma in the nerve injury+vehicle mice whereas the TNF-α concentration in control mice was 122.8 ± 5.5 pg/mL of plasma. Also, a significant increase of TNF-α was observed in the nerve injury+RSV compared to the control group. However, statistical difference was observed between RSV and vehicle groups suggesting that RSV was able to reduce inflammation after nerve injury (Figure 1E).
Similar results were observed in IL-6 biomarker analysis. The concentration of the inflammatory biomarker IL-6 was 63.6 ± 6.9 pg/mL of plasma in the nerve injury+vehicle mice whereas the IL-6 concentration in control mice was 29.6 ± 4.6 pg/mL of plasma. A slight and no significant increase of IL-6 was also observed in the nerve injury+RSV compared to the control group but this increase was lower than vehicle group, also suggesting that RSV was able to reduce inflammation after nerve injury (Figure 1F).
In order to validate the behavioral and electrophysiological data, histological analysis of sciatic nerve was performed using electron microscopy. TEM imaging demonstrated Schwann cell demyelination characterized by a disorganization of the myelin layers and axonal degeneration of sciatic nerve in the nerve injury+vehicle compared to the control group at day 4. Even if the myelin layers were less compacted in the RSV group compared to the control group, no strong demyelinating phenotype was observed in the RSV group corroborating the electrophysiology data improvement.
RSV treatment attenuated neuromotor and electrophysiological impairment in diabetic mice
After RSV efficacy study using a fast screening model, we studied the efficacy of this antioxidant in a translational and clinically relevant model, the obese diabetic mouse model.
As attended, significant body weight differences were observed from day 1 to day 62 in the two diabetic groups compared to the control group. However, a slight but significant body weight reduction was observed in the RSV treated group compared to the vehicle group suggesting an effect of RSV in glucose and fatty acid metabolism. As described in several publications, db/db diabetic mice present morbid obesity and chronic hyperglycemia leading to an increase of the body mass. Due to the strong body weight different between groups, behavioral results were normalized according individual mouse body weight.
First, coordination and equilibrium were analyzed using rotarod. Similar rotarod latency was observed in control, diabetic+vehicle and diabetic+RSV groups at baseline (4 months old). However, a progressive and significant decrease of rotarod latency was observed in the diabetic+vehicle group compared to the control group. The diabetic group presented a rotarod latency of 32.7 ± 8.2 seconds at day 60 whereas the latency of control animals remained at 139.9 ± 12.1 seconds. No significant differences were observed between diabetic+RSV and control group at day 30 and day 60. However, at day 60, a significant difference was observed between RSV and vehicle groups suggesting a protective effect of RSV in the neuromuscular impairment observed in diabetic mice (Figure 1B).
Then, the walking performance was evaluated using balance beam test. Similar balance beam cross times were observed in control, diabetic+vehicle and diabetic+RSV groups at the baseline (4 months old). However, the beam cross time was significantly increased at 14.7 ± 0.9 seconds in the diabetic+vehicle group mice whereas the cross time of control animals remained at 8.5 ± 0.4 seconds. Moreover, the diabetic+RSV group presented a significant increase of the beam cross time of 10.9 ± 0.9 seconds compared to the control group but this increase was significantly lower than vehicle treated group confirming an efficacy of RSV in diabetic induced neuromuscular impairment (Figure 2C).
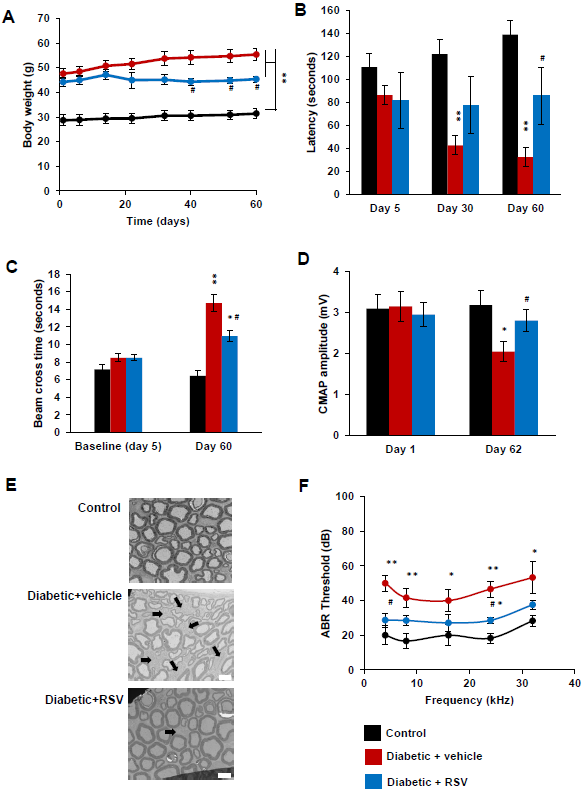
Figure 2. Peripheral neuropathy observed in diabetic mice. (A) Body weight, (B) rotarod latency, (C) balance beam cross time, (D) sciatic nerve compound muscle action potential (CMAP) amplitude, and (F) auditory brainstem responses (ABR) of control (black bars), diabetic+vehicle mice (red bars) and diabetic+RSV mice (blue bars) at the baseline (4months old) and at 6 months old (day 60-62). (E) TEM sciatic nerve imaging of control, diabetic+vehicle and diabetic+RSV. Black arrows mark non-myelinated axons. Scale bar: 5 μm. Error bars indicate SEM. Statistical tests are repeated measures two-way ANOVA test comparing groups to control (*) or vehicle (#) values. #* p<0.05, ** p<0.01, *** p<0.001. n=6
Because sciatic nerve conduction anomalies had been previously described in other publications using diabetic mice, we determine the CMAP amplitude and NCV at the baseline (4 months old) and at 6 months old (day 62).
Similar action potential amplitudes of sciatic nerve were observed in control, diabetic+vehicle and diabetic+RSV groups (around 3 mV) at baseline (4 months old). However, a significant decrease of the CMAP amplitude at 2.0 ± 0.2 mV was observed in the diabetic+vehicle group mice whereas the amplitude of control animals remained at 3.1 ± 0.3 mV.
The RSV treated group also presented a slight but no significant reduction of the CMAP amplitude at 2.9 ± 0.4 mV compared to the control group. However, the CMAP amplitude of RSV treated mice was significantly higher than vehicle treated group suggesting that RSV treatment attenuates the electrophysiological impairment of diabetic mice (Figure 2D).
As attended, no significant differences of nerve conduction velocities were observed at day 60 between control and diabetic mice (data not shown). Because several publications suggested that diabetic phenotype firstly affects Schwann cell myelination of peripheral nerve, our electrophysiology data suggests that RSV reduced diabetic neuropathy by protecting myelin structure.
RSV treatment reduced sciatic nerve histological anomalies in diabetic mice
Myelin sheath structure and axonal morphology were analyzed by TEM histology. Reduction of the myelin sheath of Schwann cell was observed in diabetic+vehicle mice, but not in the diabetic+RSV mice, at day 62 compared to control mice. This myelin sheath reduction lead to a significant increase of g-ration at 0.696 ± 0.005 for diabetic+vehicle mice whereas the diabetic+RSV and control mice presented a g-ratio mean of 0.583 ± 0.062 and 0.515 ± 0.062 respectively.
Neither axonal dead nor strong demyelination was observed in these mice. An increase of the number of nonmyelinated fibers was observed in the diabetic+vehicle group compared to the control group (black arrows). The number of non-myelinated fibers in the RSV treated group was lower than the vehicle group corroborating our previous electrophysiology data (Figure 2E).
RSV treatment reduced hearing loss in diabetic mice
Because several publications showed hearing impairment in diabetic mice. We determine the efficacy of RSV treatment in the hearing loss using ABR. ABRs are electric potentials recorded from scalp electrodes, and the first ABR wave represents the summed activity of the auditory nerve fibers contacting the inner hair cells of the cochlea.
ABR thresholds of the control and diabetic groups were not significantly different at 4 months old (data not shown). However, in the diabetic+vehicle group, the ABR thresholds significantly increased at 4 kHz, 8 kHz, 16 kHz, 24 kHz and 32 kHz at day 62 compared to the control group. In the RSV treated group, a slight increase of ABR thresholds was observed at all analyzed frequencies but this increase was only significant at 24 kHz compared to the control group (Figure 2F). Moreover, significant differences were observed between RSV and vehicle group at 2 kHz and 24 kHz suggesting a partial protective effect of RSV in the hearing impairment described in this diabetic mouse strain.
Discussion
In this study, we studied the efficacy of the antioxidant RSV in a fast screening mouse model of nerve injury.
Nerve crush induced a decrease of sciatic nerve CMAP amplitude and nerve conduction velocity, neuromuscular disorders, axonal and Schwann cell degeneration and increase of plasma inflammatory biomarkers three days after sciatic nerve injury (day 4). However, daily RSV treatment partially reduced the pathological phenotype and the neuromotor impairment in neuropathy animals induced by nerve injury.
Then, we characterized the diabetic neuropathy in an obese diabetic mouse model. We described a decrease of sciatic nerve CMAP amplitude, neuromuscular impairment, changes in sciatic nerve myelin sheath structure and hearing impairment in diabetic mice at 6 months old. However, daily RSV treatment significantly improved neuromuscular performance and nerve conduction and restored myelin structure and hearing system, suggesting a partial efficacy of RSV in neuropathy disorder induced by diabetes.
Taken together, these results confirm that: 1. Nerve injury model is a fast screening model to study the efficacy of new pharmacological candidates targeting peripheral neuropathy. 2. The db/db diabetic mouse strain is a robust and reproducible model to analyze the neuropathy and hearing disorder induced by diabetes. This model can be used to determine the efficacy of new pharmacological candidates targeting diabetic neuropathy. 3. Our data suggest than antioxidants are promising pharmacological candidates for diabetic disorders and that RSV can be used as a positive reference compound for efficacy studies.
While demyelinating peripheral nerve diseases include a large spectrum of disabling acquired and inherited diseases, the mechanisms of demyelination remain elusive. Demyelination is a cellular program that is triggered in myelinating Schwann cell upon encounter with specific signals, such as axonal injury in a trauma. These triggering signals are transduced in the cell, leading to the recruitment of phosphorylated c-JUN in the nucleus [12,13]. This gene participates in the demyelination program in the nucleus and mediates a protective effect against loss of sensory axons [14]. Moreover, peripheral nerve demyelination can be initiated by various mechanisms, including nerve injury, metabolic alterations, and gene mutations. Such heterogeneity has hindered the search for a common treatment for these peripheral neuropathies.
In this study we used the db/db mouse strain. This strain is used to model phases I to III of diabetes type II and obesity. Mice homozygous for the diabetes spontaneous mutation (Leprdb) manifest morbid obesity, chronic hyperglycemia, pancreatic beta cell atrophy and become hypoinsulinemic. Obesity starts at 3 to 4 weeks of age [15,16]. Elevated plasma insulin begins at 10 to 14 days and elevated blood sugar at 4 to 8 weeks. Homozygous mice are polyphagic, polydipsic, and polyuric. The severity of disease on BKS background leads to uncontrolled rise in blood sugar, severe depletion of insulin-producing beta-cells of the pancreatic islets, peripheral neuropathy, myocardial disease and death by 10 months of age. Exogenous insulin fails to control blood glucose levels and gluconeogenic enzyme activity increases. Wound healing is delayed, and metabolic efficiency is increased. In rodent and humans, diabetic peripheral neuropathy is deeply linked to oxidative stress and mitochondrial dysfunctions [17]. However, the causes of diabetic peripheral nerve defect this neuropathy remain poorly studied. Diabetic peripheral neuropathy in humans is characterized by a chronic demyelination [9].
ROS are very unstable species, reacting with other molecules and thereby damaging proteins, lipids and DNA [18]. In this way, ROS generated by mitochondria are hypothesized to damage key mitochondrial components, such as mitochondrial DNA (mtDNA), mitochondrial membranes, respiratory chain proteins, and nuclear DNA that affect mitochondrial function [19]. ROS-induced damage has the potential to alter electron transport chain function, decrease the efficiency of ATP production and increase inflammation and NADPH oxidase (Figure 3). Moreover, several studies proposed the free radical theory of ageing, which suggested that free radicals can affect and are possibly the cause of the ageing process of animals [20]. This creates a “vicious cycle” in which ROS production from the mitochondrial electron transport chain is able to cause damage to the mtDNA found in close proximity. Because mtDNA encodes the majority of mitochondrial proteins, errors in gene expression of mtDNA may result in dysfunctional mitochondrial subunits. Dysfunctional mitochondria are then thought to contribute to further ROS leakage due to their inefficiency, which could then exacerbate mtDNA damage in a continuing vicious cycle [21,22].
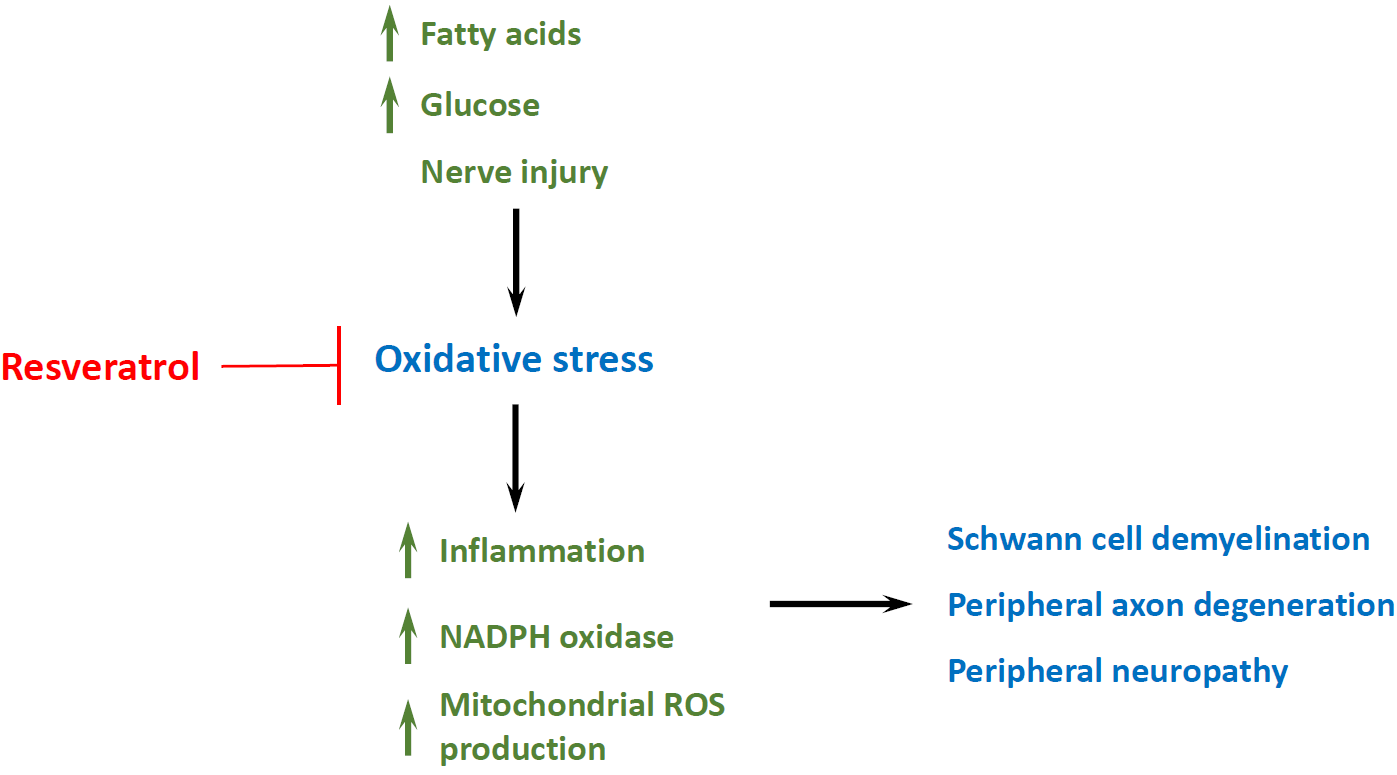
Figure 3. Schematic representation of the oxidative stress pathway leading to peripheral nervous system demyelination and axonal degeneration. Resveratrol reduces cellular oxidative stress attenuating peripheral neuropathy.
Increasing number of publications demonstrated that mitochondrial ROS production plays a key role in peripheral neuropathy by Schwann cell demyelination and axon apoptotic pathway activation. Major ROS production pathway includes oxidative phosphorylation dysfunction, nuclear transcription factors activation, mtDNA damage, decreased anti-ROS enzyme activity, etc. Even if increasing number of publications suggest that RSV treatment attenuates peripheral neuropathy and ameliorates histological functions [23-25], the effect of antioxidants supplementation, such as RSV, in the prevention of peripheral neuropathy and other pathologies is presently controversial and inconclusive. Development of effective technique for antioxidant therapy in neuromotor disorders requires an understanding of myelinating Schwann cell and axon oxidative stress mechanisms underlying the peripheral neuropathy that may overcome this obstacle by opening doors to new treatment options.
References
2. Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nature Reviews Neuroscience. 2005 Sep;6(9):683-90.
3. Richard H. AC Peripheral neuropathy. BMJ. 2002;23:466-9.
4. Zochodne DW. Diabetic polyneuropathy: an update. Current opinion in neurology. 2008 Oct 1;21(5):527-33.
5. Zenker J, Ziegler D, Chrast R. Novel pathogenic pathways in diabetic neuropathy. Trends in Neurosciences. 2013 Aug 1;36(8):439-49.
6. Pham K, Gupta R. Understanding the mechanisms of entrapment neuropathies. Neurosurgical Focus. 2009 Feb 1;26(2):E7.
7. Harati Y. Diabetic peripheral neuropathies. Annals of Internal Medicine. 1987 Oct 1;107(4):546-59.
8. Sugimoto K, Murakawa Y, Sima AA. Diabetic neuropathy–a continuing enigma. Diabetes/Metabolism Research and Reviews. 2000 Nov;16(6):408-33.
9. Malik RA. Pathology of human diabetic neuropathy. InHandbook of clinical neurology 2014 Jan 1 (Vol. 126, pp. 249-259). Elsevier.
10. De La Lastra CA, Villegas I. Resveratrol as an antioxidant and pro-oxidant agent: mechanisms and clinical implications. Biochemical Society Transactions. 2007 Nov;35(Pt 5):1156-60.
11. Shaito A, Posadino AM, Younes N, Hasan H, Halabi S, Alhababi D, Al-Mohannadi A, et.al. Potential adverse effects of resveratrol: a literature review. International Journal of Molecular Sciences. 2020 Jan;21(6):2084.
12. Lee HJ, Shin YK, Park HT. Mitogen activated protein kinase family proteins and c-jun signaling in injury-induced Schwann cell plasticity. Experimental Neurobiology. 2014 Jun 1;23(2):130-7.
13. Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, et.al. c-Jun is a negative regulator of myelination. The Journal of Cell Biology. 2008 May 19;181(4):625-37.
14. Hantke J, Carty L, Wagstaff LJ, Turmaine M, Wilton DK, Quintes S, Koltzenburg M, et.al. c-Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain. 2014 Nov 1;137(11):2922-37.
15. Robertson DM, Sima AA. Diabetic neuropathy in the mutant mouse [C57BL/ks (db/db)]: a morphometric study. Diabetes. 1980 Jan 1;29(1):60-7.
16. Hamilton RT, Bhattacharya A, Walsh ME, Shi Y, Wei R, Zhang Y, Rodriguez KA, et.al. Elevated protein carbonylation, and misfolding in sciatic nerve from db/db and Sod1-/- mice: plausible link between oxidative stress and demyelination. PLoS One. 2013 Jun 4;8(6):e65725.
17. Feldman EL. Oxidative stress and diabetic neuropathy: a new understanding of an old problem. The Journal of Clinical Investigation. 2003 Feb 15;111(4):431-3.
18. Jones DP. Radical-free biology of oxidative stress. American Journal of Physiology-Cell Physiology. 2008 Oct;295(4):C849-68.
19. Seidman MD, Ahmad N, Joshi D, Seidman J, Thawani S, Quirk WS. Age-related hearing loss and its association with reactive oxygen species and mitochondrial DNA damage. Acta Oto-Laryngologica. 2004 Apr 1;124(0):16- 24.
20. Barja G. Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxidants & Redox Signaling. 2013 Oct 20;19(12):1420-45.
21. Bandy B, Davison AJ. Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging?. Free Radical Biology and Medicine. 1990 Jan 1;8(6):523-39.
22. Kandola K, Bowman A, Birch-Machin MA. Oxidative stress–a key emerging impact factor in health, ageing, lifestyle and aesthetics. International Journal of Cosmetic Science. 2015 Dec;37:1-8.
23. Zhang J, Ren J, Liu Y, Huang D, Lu L. Resveratrol regulates the recovery of rat sciatic nerve crush injury by promoting the autophagy of Schwann cells. Life Sciences. 2020 Jun 10:117959.
24. Ding Z, Cao J, Shen Y, Zou Y, Yang X, Zhou W, Guo Q, Huang C. Resveratrol promotes nerve regeneration via activation of p300 acetyltransferase-mediated VEGF signaling in a rat model of sciatic nerve crush injury. Frontiers in Neuroscience. 2018 May 23;12:341.
25. San Cheang W, Wong WT, Wang L, Cheng CK, Lau CW, Ma RC, et.al. Resveratrol ameliorates endothelial dysfunction in diabetic and obese mice through sirtuin 1 and peroxisome proliferator-activated receptor d. Pharmacological Research. 2019 Jan 1;139:384-94.