Abstract
The pancreatic islets of Langerhans contain a minority of endocrine cells that contain more than one hormone: predominantly combinations of glucagon, insulin and somatostatin. A recent paper from our laboratory examined the ontogeny of such cells in the human pancreas and found that they persisted throughout the lifespan but altered in relative abundance with age. Glucagon/insulin bi-hormonal cell number significantly increased with age whilst insulin/somatostatin and glucagon/somatostatin cells significantly decreased. Building on that study here we explore the possible origins and physiological role of bi-hormonal cells within the endocrine pancreas. During pancreas development in utero mono-hormonal endocrine cell lineages are defined by distinct signatures of transcription factor expression. Insufficient or inappropriately timed expression in sub-populations of endocrine cell progenitors may fail to suppress genes normally restricted to other endocrine cell types, resulting in residual populations of bi-hormonal cells. These have been identified postnatally by single cell transcriptomic or proteomic analysis and do not appear to have a metabolic role distinct from that of mono-hormonal cells. However, during hyperglycemic stress bi-hormonal cell sub-populations can proliferate and transdifferentiate into new beta-cells capable of glucose-stimulated insulin release. Transdifferentiation is reversible and in both type 1 and 2 diabetes some beta-cells can dedifferentiate to form hormone-null cells or alpha-cells. Thus, current evidence suggests that diverse phenotypic pools of endocrine cell phenotypes are retained within the islets into adult life that can undergo lineage changes in response to metabolic demand. There is preliminary evidence that these pathways can be therapeutically manipulated.
Keywords
Pancreas, Islets of Langerhans, Bi-hormonal, Alpha-cell, Beta-cell, Delta-cell, Transdifferentiation, Dedifferentiation
Introduction
The pancreatic islets of Langerhans contain distinct endocrine hormone populations of which the major phenotypes are insulin-secreting beta-cells, glucagon-secreting alpha-cells, and somatostatin-secreting delta-cells [1]. Within the human pancreas these are present in the approximate ratio of 60:30:10% respectively. Other minority endocrine cell types are also present contributing 1-2% of the islets including pancreatic polypeptide secreting cells and ghrelin-secreting epsilon cells. However, a minority of each endocrine cell type appear to be bi-hormonal in nature. We recently published an analysis of the presence and ontogeny of bi-hormonal islet cells within the human pancreas across the lifespan in individuals without diabetes utilizing immunofluorescence co-localization [2]. Bi-hormonal cells were observed at all ages from birth until the eighth decade with the most abundant cell phenotype containing both glucagon and insulin, representing 3% of all islet cells. Glucagon/somatostatin, and insulin/somatostatin bi-hormonal cells were also observed but at lower abundance. We found that glucagon/insulin bi-hormonal cell number significantly increased with age whilst insulin/somatostatin and glucagon/somatostatin cells significantly decreased. While the study showed that bi-hormonal cells are present within human islets throughout life it did not shed light on the origins or physiological potential of these cell populations, or how they might change during metabolic stress or diabetes. This commentary offers some perspectives.
Bi-hormonal Cell Presence in the Healthy Pancreas
Studies by Kang et al. [3], used both single cell RNA sequencing (scRNA-seq) and single cell nucleus RNA sequencing to identify five sub-clusters of glucagon-expressing cells within isolated islets from healthy human donors. One cluster co-expressed glucagon/insulin and could differentiate into either glucagon or insulin mono-hormonal cells in vitro. When maintained as islet grafts in vivo though, these cells showed unidirectional plasticity to become beta-cells only. A specific gene expression signature was identified during the alpha- to beta-cell transdifferentiation including genes regulating calcium-channel function, long non-coding RNAs, DNA-binding proteins, and histone deacetylases; the latter suggesting epigenetic modification of histone structure to support the repression of genes typical of alpha-cells, such as the processing enzymes regulating the relative synthesis of glucagon versus glucagon-related peptide-1 (GLP-1), and the up-regulation of genes required for glucose-stimulated insulin secretion (GSIS).
Both Saikia et al. [4] and Elgamal et al. [5], identified distinct alpha-cell sub-clusters expressing both insulin and glucagon within human islets. When scRNA-seq was used to document the transcriptome of human pancreatic slices from non-diabetic subjects, multiple sub-clusters of insulin-expressing cells were found, with one of these co-expressing glucagon and TMSB10, a gene associated with cytoskeletal reorganization and cell motility [6]. If these cells contribute towards alpha- to beta-cell transdifferentiation this might suggest an associated anatomical restructuring of islet cell connectivity within the islets. Yu et al. [7] used co-expression of specific islet endocrine lineage markers in normal mouse pancreas to study the transcriptomes of rare bi-hormonal cell populations. The most frequent of these were glucagon/insulin and glucagon/pancreatic polypeptide (PPY) phenotypes, both of which tended to have a functional transcriptome similar to alpha-cells. However, a shortcoming of transcriptomic analysis alone is that some dispersed human alpha-cells contained a high insulin mRNA content that was not translated into insulin [8], reinforcing the importance of considering both transcriptome and proteome when assigning cell phenotype. A deep proteomic analysis of single cells within sections of adult human pancreas identified twelve endocrine sub-types, including those with cell surface markers for both alpha/beta-cells, delta/beta-cells, and alpha/delta-cells [9]. The highest frequency of alpha/beta-cells was found in a sub-population of small sized islets. Functional proteomic analysis revealed that both beta-cells and some beta/delta-cells were engaged in TCA cycle, fatty acid and glucose metabolism, suggesting that the bi-hormonal cells exhibited beta-cell-like function. Alpha/beta-hybrid cells expressed progenitor cell and cell cycle proliferation markers suggesting the potential for developmental plasticity. These data suggest that in the normal adult pancreas there are bi-hormonal cell sub-populations that are both metabolically functional and have trans-lineage plastic potential.
Bi-hormonal Cells as a Residue of Fetal Development
One explanation of the presence of bi-hormonal cells postnatally is that they represent a vestige of the developmental pathways from which islet endocrine cell lineages derived in utero. Pancreas development commences with the expression of the transcription factor pancreatic and duodenal homeobox 1 (Pdx1) in the foregut dorsal endoderm in two ventrolateral regions, which then undergo morphogenesis to create pancreatic rudiments [10]. An orchestrated cascade of transcription factor expression follows to convert primitive endoderm into the dorsal and ventral pancreatic buds [11]. Around embryonic day 9.5 to 10.5 in the mouse fusion of the dorsal and ventral pancreas rudiments occurs, and a branching morphogenesis follows to create a ductal tree from which both exocrine and endocrine cell lineages are generated. Beta-and other endocrine cell progenitors develop from the mid-region epithelium of each primitive duct and, accompanied by endothelial cells that produce the islet basement membrane, migrate into the parenchyma and self-assemble into individual islets, each with a specialist vascular bed (Figure 1). Glucagon/insulin bi-hormonal cells are seen within the human fetal pancreas at weeks 6-7 of gestation but disappear during later islet maturation [12]. This ontogeny is supported by studies in vitro of the differentiation of alpha and beta-cells from human embryonic stem cells where insulin/glucagon bi-hormonal cells appear coincident with the appearance of the endocrine cell populations [13]. The functional maturation of beta-cells in vivo occurs prior to birth in human but between birth and weaning in the mouse [14].
Single cell RNAseq analysis of the human fetal pancreas showed that bi-hormonal cells have a transcriptional profile more similar to alpha-cells than beta-cells [7]. Beta-like cells generated in vitro from human embryonic stem cells contain a bi-hormonal cell population twice that of insulin mono-hormonal cells [15]. However, most of these cells were clustered around alpha-cells and expressed very low levels of insulin. Despite the apparent absence of a distinct functional transcriptional profile in fetal bi-hormonal cells there is substantial plastic potential, and fetal islet cells committed to the synthesis of one hormone can give rise of different mono-hormonal cells in the adult pancreas [16]. The phenotype of bi-hormonal cells may represent a weakened timing or magnitude of expression of transcription factors that are signatures for each defined endocrine cell type. During islet differentiation within the fetus most transcription factors both induce a dominant endocrine cell phenotype while also suppressing transcription factors defining other endocrine cell types. An example of this is a sub-optimal expression of Foxo1in beta-cells which results in cells that are multi-hormonal [17]. Thus, a residual population of bi-hormonal cells in the islets following birth may simply represent the transcriptional outliers of the endocrine lineage separation process in utero that did not become definitive mono-hormonal cells.
Bi-hormonal Cells as a Regenerative Mechanism
Why then might a bi-hormonal cell population be retained into adult life? One possibility is that they represent a regenerative reserve of endocrine cells with plastic lineage capacity to address a sub-optimal endocrine cell balance during times of metabolic stress. The lineage commitment of both alpha- and beta-cells is relatively epigenetically shallow and can be altered by the experimental ectopic expression of a relatively small number of transcription factors. Human donor islets transfected with the transcription factors PDX1 and MAF BZIP Transcription Factor A (MAFA), which contribute to the beta-cell phenotype, underwent alpha- to beta-cell transdifferentiation following transplant into diabetic mice, as did alpha-cells depleted of ARX expression, a key gene necessary for alpha-cell function [18]. Over-expression of the beta-cell transcription factor Paired Box 4 (Pax4) caused the conversion of alpha-cells into insulin-producing cells in a mouse model [19], with similar findings following transfection of human islets from subjects with type 2 diabetes [20]. Hence, naturally occurring bi-hormonal cells may be primed for a rapid phenotypic conversion as a result of prolonged hyperglycemic stress.
We observed that bi-hormonal cells are often located towards the rim of both mouse and human islets [2] which has been proposed as a niche for resident beta-cell progenitors [21,22]. It is possible, therefore, that this also represents a preferred location for endocrine cell transdifferentiation. Since the islet rim is an area of extensive juxtaposition of alpha-, beta-, delta- and epsilon-cells it represents a site of high paracrine cell interactions, for instance the availability of alpha-cell-derived GLP-1.
A pool of resident, phenotypically plastic bi-hormonal cells would be advantageous as the regenerative capacity of beta-cells by cell replication alone is extremely low both in humans and in animal models beyond early childhood [23,24]. There remains a sub-population of progenitor beta-cells within islets following birth, which express insulin but exhibit poor GSIS, due to a paucity of glucose receptor-2 (GLUT2) expression [25,26], their abundance declines rapidly after puberty [27]. The possibility that these are derived from bi-hormonal cells seems unlikely since they are transcriptionally distinct by scRNA-seq, at least in the mouse pancreas [28]. Similarly, other sub-populations of insulin-expressing cells such as the hub beta-cells that coordinate pulsing of intracellular calcium levels and first-phase insulin release are transcriptionally different from bi-hormonal cells [29].
In the face of the poor ability of beta-cells to proliferate in adult life a functional increase in insulin availability in response to insulin resistance might be achieved by lineage plasticity of islet endocrine cells, and an increase in bi-hormonal cell abundance (Figure 1). In support of this concept the number of insulin-immunoreactive cells also containing either glucagon or somatostatin in human islets was 2-3-fold greater in donors with type 2 diabetes than in metabolically healthy controls [30–32]. This may boost insulin secretion capacity and counteract insulin resistance in type 2 diabetes [17]. Arguing against such a direct relationship with insulin demand was a lack of correlation seen between bi-hormonal cell number and basal or first-phase insulin secretion, or the total insulin secretion during a hyperglycemic clamp [33]. This would suggest that bi-hormonal glucagon/insulin cells are not themselves likely to greatly contribute to GSIS but are more likely to represent an intermediate step in a more complete transdifferentiation into functional beta-cells under metabolic stress.
To what extent is transdifferentiation to beta-cells possible in response to hyperglycemic stress alone? Following a rapid induction of hyperglycemia in mice using the beta-cell toxin, streptozotocin, cell lineage tracing demonstrated a prompt increase in glucagon/insulin bi-hormonal cells, but only around 10% of these became mono-hormonal insulin-expressing cells over a period of three weeks [34]. This equates to approximately 2% of islet alpha-cells that were capable of full transdifferentiation, but this was sufficient to enable a partial recovery of beta-cell mass. Pharmacologic inhibition of the glucagon receptor activation was able to reverse diabetes in type 1 diabetic mice with an estimated 15% of new beta-cells being derived from alpha-cells through transdifferentiation assessed by lineage tracking [35]. Transcriptional and epigenetic changes in islet endocrine cells have been followed longitudinally during the induction of diabetes in mice exposed to a high fat diet [36]. Intercellular communication between islet cells, and between islet cells and the vascular bed or with macrophages were all modified, and the population of alpha- and delta-cells both relatively declined compared to beta-cells. The presence of several bi-hormonal cell populations changed in abundance during the development of hyperglycemia, including insulin/somatostatin and glucagon/somatostatin cells. Two populations of alpha-cells were identified: one retaining strong alpha-cell-related functional activity and the other showing reduced glucagon expression and a more beta-cell transitional phenotype. Three populations of delta-cells were found in the progression towards diabetes: the first retaining a strong delta-cell transcriptional profile, a second showing a transitional profile towards beta-cells with both INS1 and INS2 expression, and the third beginning to express alpha-cell transcriptional markers. These findings highlight multiple islet cell transcriptional responses when faced with hyperglycemic and hyperlipidemic stress with sub-populations of alpha- and delta-cells increasingly adopting beta-cell characteristics.
However, adult beta-cells can be experimentally manipulated to re-enter the proliferative cycle outside of metabolic stress. Adult human beta-cells reinitiated proliferation both in vitro and when grafted into mice in vivo following exposure to the Dual specificity tyrosine-phosphorylation-regulated kinase 1A (DYRK1A) inhibitor, harmine, together with a GLP-1 receptor agonist [37]. Subsequent analysis showed that DYRK1A inhibitors activated a sub-population of alpha-cells to proliferate, adopt a glucagon/insulin bi-hormonal phenotype, and undergo transdifferentiation to become beta-cells capable of GSIS [38]. A variety of other pharmacological interventions have also been found to induce beta-cell mass through transdifferentiation (Table 1). Alpha- to beta-cell transdifferentiation was enhanced in human islets by exposure to GLP-1 alone [4] and in rodent islets following administration of gamma aminobutyric acid (GABA) [39], which resulted in a downregulation of the transcription factor Aristaless Related Homeobox (Arx) in alpha-cells. Serotonin treatment increased the percentage of glucagon/insulin bi-hormonal cells along with beta-cell-specific transcription factors such as NK6 Homeobox 1 (Nkx6.1) in both isolated mouse and human islets, and in vivo in mice fed a high fat diet as a model for type 2 diabetes [40]. GLP-1, GABA and serotonin are each paracrine trophic beta-cell factors released from alpha-cells (GLP-1) or beta-cells (GABA and serotonin) during hyperglycemic stress [41]. When considering a possible therapeutic strategy, the SGLT2 inhibitor, dapagliflozin, used in the control of T2D also promoted alpha- to beta-cell transdifferentiation in diabetic mice [42].
Islet cell trandifferentiation to enhance the beta-cell population is not limited to alpha-cells since a bi-hormonal phenotype expressing both somatostatin and insulin was identified during induced transdifferentiation of delta- to beta-cells following the destruction of beta-cells in the zebrafish [43] (Figure 1). Similarly, an experimental loss of beta-cells in pre-weaning mice caused a subsequent regeneration by transdifferentiation from delta-cells, but this pathway was reduced substantially as the animals became adult when alpha- to beta-cell transdifferentiation become more frequent [44]. The somatostatin/insulin bi-hormonal cell population observed in early life may be transient and derived from pancreatic duct epithelium since Neurogenin-3 (Ngn3)-expressing ductal endocrine progenitors’ express somatostatin and subsequently transdifferentiate into beta-cells [45].
A negative correlation exists between increasing age and islet cellular plasticity [33,46] which, if bi-hormonal cells represent an intermediary mechanism towards beta-cell expansion might reflect their decreased abundance. The relative abundance of alpha-cells in human islets increases with age until around 30 years, whilst over the lifespan delta-cell presence decreases with age [31]. We found that the proportion of beta-cells per islet increased during childhood to reach adult values around the time of puberty, but thereafter alpha to beta-cell ratio remained constant [2]. Histological analysis has shown that beta-cell mass increases thirty-fold between birth and adulthood [47], with the growth velocity being greatest between birth and 2 years age [33], after which the beta-cells become proliferatively quiescent [48]. With respect to bi-hormonal cell ontogeny, as stated previously, we found a negative relationship between insulin/somatostatin and glucagon/somatostatin bi-hormonal cells in the human pancreas, but glucagon/insulin cell abundance increased with age [2]. This suggests that the decrease in islet cell regenerative capacity with age is not directly attributable to a loss bi-hormonal cell
Bi-hormonal Cells Reflect Dedifferentiation for Beta-cell Survival
An increase in bi-hormonal cell abundance during hyperglycemia could represent an attempt to increase insulin secretion capacity, but it might also represent a dedifferentiation of beta-cells to an alpha-cell phenotype as a result of metabolic stress, as seen in type 2 diabetes [17] (Figure 1). Beta-cell dedifferentiation has also been observed as part of normal aging [46]. Beta-cell mass is lost during type 2 diabetes, in part through apoptosis in response to gluco- and lipotoxicity. However, the rate of apoptosis observed in animal models is insufficient to fully account for the beta-cell loss. It is thought that the predominant mechanism to account for loss of beta-cells is dedifferentiation either to a hormone-null, degranulated progenitor endocrine-like state [17], or a transdifferentiation to adopt an alpha-cell phenotype [49]. A similar dedifferentiation of beta-cells has been reported to occur in type 1 diabetes also and may represent a survival mechanism to avoid autoimmune destruction, especially if the phenotypic change is potentially reversible [50].
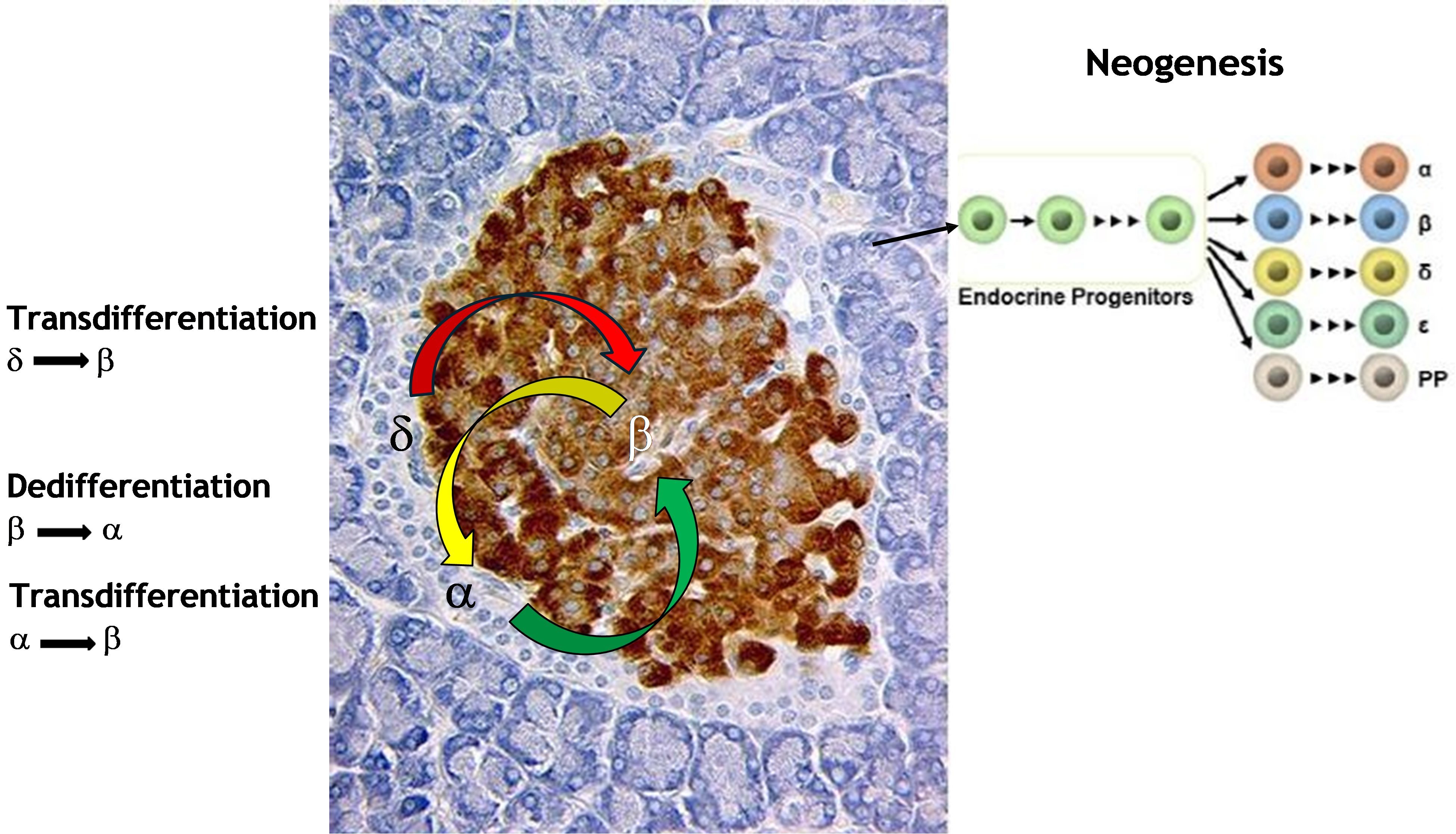
Figure 1. Neogenesis of islet endocrine cells from multipotent progenitors within the pancreatic ducts occurs during fetal and neonatal life resulting in lineage commitment into alpha- (α), beta- (β), delta- (δ), epsilon (ε) and pancreatic polypeptide cells (PP). Transdifferentiation can occur in adulthood by conversion of alpha- or delta-cells either directly or via a bi-hormonal intermediary phenotype to become phenotypic beta-cells. During type 2 diabetes beta-cells may dedifferentiate under hyperglycemic stress to become phenotypic alpha-cells.
Beta-cell dedifferentiation is associated with a relative loss of expression of genes defining beta-cells, such as the transcription factors Pdx1, Nkx6.1 and MafA [51,52]; the appearance of lineage precursor-associated gene expression such as Ngn3, Nanog and aldehyde dehydrogenase 1 family member A3 (Aldh1a3) [53], and the expression of genes associated with alpha-cells that would normally be repressed in beta-cells such as proglucagon and Arx [54] (Table 1). Some beta-cells may progress to a hormone-negative phenotype, others transdifferentiating into an alpha-cell phenotype, and some simply becoming bi-hormonal, depending on the extent and duration of metabolic stress. Presently, there is a lack of a distinct transcriptional signature for bi-hormonal cells that can distinguish between alpha-cells transdifferentiating to become beta-cell versus beta-cells dedifferentiating and adopting an alpha-cell phenotype.
Importantly for therapeutic strategies the beta-cell dedifferentiated state appears to be reversible. Early intervention in type 2 diabetes with insulin together with metformin improved beta-cell function with long-term diabetes remission [55]. In the db/db mouse model of diabetes calorie restriction was able to reverse beta-cell dedifferentiation [56], while cellular therapy though the grafting of umbilical cord-derived mesenchymal stem cells was also able to reverse engineer dedifferentiation through the re-expression of transcription factors such as Pdx1 [57].
|
|
Transdifferentiation α→β |
Dedifferentiation β→α |
|---|---|---|
|
Experimental models |
β-cell loss (STZ, Diphtheria toxin) |
High fat diet, hyperglycaemia |
|
Gene and protein markers |
Proinsulin, PC1/3, MafA, GLUT2 |
Proglucagon, Arx, Aldh1a3 |
|
Lineage transition triggers |
GLP-1, GABA, Serotonin SGLT2 inhibition DYRK1A inhibition |
Type 2 diabetes |
|
Functional capacity |
Immature first phase insulin release, reduced insulin content |
Glucose-sensitive glucagon release |
Conclusion
Islet bi-hormonal cells are present as a sub-population of endocrine cells throughout life, first appearing as part of the lineage development of the endocrine pancreas during fetal organogenesis. There is extensive phenotypic diversity throughout the islet endocrine cell population which persists into adulthood. While there are limited evidence that bi-hormonal cells themselves add an additional element of glycemic control compared to mono-hormonal islet cells, they can undergo transdifferentiation to become functional beta-cells under conditions of hyperglycemic stress. This involves a transcriptional realignment whereby beta-cell-specific genes are upregulated and alpha- or delta-cell associated genes are repressed and are under epigenetic control. Some bi-hormonal cell populations are also proliferative, thereby additionally expanding the potential for transdifferentiation. Transdifferentiation can be enhanced in response to therapeutic agents such as GLP-1 receptor agonists or serotonin. However, bi-hormonal cells might also result from a dedifferentiation of beta-cells during diabetes to avoid cellular stress and death. The balance between a capacity for transdifferentiation to yield new beta-cells and a loss of beta-cell function through dedifferentiation is not understood but is likely to be influenced by the degree and duration of metabolic stress. Can the transdifferentiation of bi-hormonal cells be considered as a viable therapeutic option to prevent or control diabetes? It may already be contributing to the actions of commonly used drugs for glycemic control such as GLP-1 receptor agonists or metformin.
Key information is still lacking to enable transdifferentiation of non-beta islet cells into functional beta-cells in vivo as a potential therapeutic strategy to delay or prevent the onset of diabetes. Firstly, it is unclear the extent to which transdifferentiation gives rise to fully functional, glucose-sensitive beta-cells. Whilst some information can be gleaned from transcriptomic and proteomic analysis of single transdifferentiated cells in situ, specific molecular biomarkers will be needed to bulk separate transdifferentiated from ‘wild type’ beta-cells for detailed kinetic analysis of glucose-stimulated insulin secretion. Secondly, whilst a number of small molecules have been suggested to initiate alpha- to beta-cell transdifferentiation many of these are repurposed drugs with widespread additional actions throughout the body. Effective dosage in enabling islet cell transdifferentiation versus other actions will require detailed pharmacokinetic analysis in a standardized animal model. However, the accumulating findings that islet cells can adopt a spectrum of overlapping lineage phenotypes provides impetus for further in-depth study.
Acknowledgements
For research performed by the author thanks are extended to the Canadian Institutes of Health Research, the Lawson Foundation and Lawson Research Institute for financial support.
References
2. Hahm J, Kumar D, Andrade JAF, Arany E, Hill DJ. Bi-Hormonal Endocrine Cell Presence Within the Islets of Langerhans of the Human Pancreas Throughout Life. Cells. 2025 Jan 1;14(1):34.
3. Kang RB, Lee J, Varela M, Li Y, Rosselot C, Zhang T, et al. Human pancreatic α-cell heterogeneity and trajectory inference analysis using integrated single cell-and single nucleus-RNA sequencing platforms. BioRxiv. 2023 Nov 19:2023–11.
4. Saikia M, Holter MM, Donahue LR, Lee IS, Zheng QC, Wise JL, et al. GLP-1 receptor signaling increases PCSK1 and β cell features in human α cells. JCI insight. 2021 Feb 8;6(3):e141851.
5. Elgamal RM, Kudtarkar P, Melton RL, Mummey HM, Benaglio P, Okino ML, et al. An integrated map of cell type–specific gene expression in pancreatic islets. Diabetes. 2023 Nov 1;72(11):1719–28.
6. Doke M, Álvarez-Cubela S, Klein D, Altilio I, Schulz J, Goncalves LM, et al. Dynamic scRNA-seq of live human pancreatic slices reveals functional endocrine cell neogenesis through an intermediate ducto-acinar stage. Cell Metabolism. 2023 Nov 7;35(11):1944–60.
7. Yu XX, Peng P, Wang YN, He MY, He S, Jin CT, et al. Comprehensive Characterization of Bihormonal Cells and Endocrine Cell Lineages in Mammalian Pancreatic Islets. Advanced Science. 2025 May;29:e16326.
8. Blodgett DM, Nowosielska A, Afik S, Pechhold S, Cura AJ, Kennedy NJ, et al. Novel observations from next-generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes. 2015 Sep 1;64(9):3172–81.
9. Mardamshina M, Dorka N, Thielert M, Björklund F, Navarro FB, Casals AM, et al. Multiplexed Deep Visual Proteomics Unveils Spatial Heterogeneity and Rare Endocrine States in Human Adult Pancreatic Islets. BioRxiv. 2025 Apr 27:2025–04.
10. Nikolova G, Lammert E. Interdependent development of blood vessels and organs. Cell and Tissue Research. 2003 Oct;314(1):33–42.
11. Lammert E, Cleaver O, Melton D. Induction of pancreatic differentiation by signals from blood vessels. Science. 2001 Oct 19;294(5542):564–7.
12. Villalba A, Gitton Y, Aiello V, Toupin M, Mazaud-Guittot S, Chédotal A, et al. Imaging human pancreatic endocrinogenesis during early prenatal life. Diabetes. 2025 Mar 1;74(3):368–75.
13. Mar S, Filatov E, Sasaki S, Mojibian M, Zhang D, Yang A, et al. Tracking Insulin-and Glucagon-Expressing Cells In Vitro and In Vivo Using a Double-Reporter Human Embryonic Stem Cell Line. Diabetes. 2025 Feb 1;74(2):188–98.
14. Trudeau JD, Dutz JP, Arany E, Hill DJ, Fieldus WE, Finegood DT. Neonatal beta-cell apoptosis: a trigger for autoimmune diabetes? Diabetes. 2000 Jan 1;49(1):1–7.
15. Zhu H, Wang G, Nguyen-Ngoc KV, Kim D, Miller M, Goss G, et al. Understanding cell fate acquisition in stem-cell-derived pancreatic islets using single-cell multiome-inferred regulomes. Developmental Cell. 2023 May 8;58(9):727–43.
16. Perez-Frances M, Abate MV, Baronnier D, Scherer PE, Fujitani Y, Thorel F, et al. Adult pancreatic islet endocrine cells emerge as fetal hormone-expressing cells. Cell Reports. 2022 Feb 15;38(7):110377.
17. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. 2012 Sep 14;150(6):1223–34.
18. Furuyama K, Chera S, van Gurp L, Oropeza D, Ghila L, Damond N, et al. Diabetes relief in mice by glucose-sensing insulin-secreting human α-cells. Nature. 2019 Mar 7;567(7746):43–8.
19. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, et al The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and subsequently β cells. Cell. 2009 Aug 7;138(3):449–62.
20. Zhang Y, Parajuli KR, Fonseca VA, Wu H. PAX4 gene delivery improves β-cell function in human islets of Type II diabetes. Regenerative Medicine. 2024 May 3;19(5):239–46.
21. van der Meulen T, Mawla AM, DiGruccio MR, Adams MW, Nies V, Dólleman S, et al. Virgin beta cells persist throughout life at a neogenic niche within pancreatic islets. Cell Metabolism. 2017 Apr 4;25(4):911–26.
22. Lee S, Zhang J, Saravanakumar S, Flisher MF, Grimm DR, van der Meulen T, et al. Virgin β-cells at the neogenic niche proliferate normally and mature slowly. Diabetes. 2021 May 1;70(5):1070–83.
23. Gregg BE, Moore PC, Demozay D, Hall BA, Li M, Husain A, et al. Formation of a human β-cell population within pancreatic islets is set early in life. The Journal of Clinical Endocrinology & Metabolism. 2012 Sep 1;97(9):3197–206.
24. Tudurí E, Soriano S, Almagro L, Montanya E, Alonso-Magdalena P, Nadal Á, et al. The pancreatic β-cell in ageing: Implications in age-related diabetes. Ageing Research Reviews. 2022 Sep 1; 80:101674.
25. Smukler SR, Arntfield ME, Razavi R, Bikopoulos G, Karpowicz P, Seaberg R, et al. The adult mouse and human pancreas contain rare multipotent stem cells that express insulin. Cell Stem Cell. 2011 Mar 4;8(3):281–93.
26. Beamish CA, Strutt BJ, Arany EJ, Hill DJ. Insulin-positive, Glut2-low cells present within mouse pancreas exhibit lineage plasticity and are enriched within extra-islet endocrine cell clusters. Islets. 2016 Apr 18;8(3):65–82.
27. Beamish CA, Mehta S, Strutt BJ, Chakrabarti S, Hara M, Hill DJ. Decrease in Ins+ Glut2 LO β-cells with advancing age in mouse and human pancreas. Journal of Endocrinology. 2017;233(3):229–41.
28. Feng Y, Qiu WL, Yu XX, Zhang Y, He MY, Li LC, et al. Characterizing pancreatic β-cell heterogeneity in the streptozotocin model by single-cell transcriptomic analysis. Molecular Metabolism. 2020 Jul 1; 37:100982.
29. Johnston NR, Mitchell RK, Haythorne E, Pessoa MP, Semplici F, Ferrer J, et al. Beta cell hubs dictate pancreatic islet responses to glucose. Cell Metabolism. 2016 Sep 13;24(3):389–401.
30. Butler AE, Campbell-Thompson M, Gurlo T, Dawson DW, Atkinson M, Butler PC. Marked expansion of exocrine and endocrine pancreas with incretin therapy in humans with increased exocrine pancreas dysplasia and the potential for glucagon-producing neuroendocrine tumors. Diabetes. 2013 Jul 1;62(7):2595–604.
31. Toledo MP, Xie G, Wang YJ. Comprehensive characterization of islet remodeling in development and in diabetes using mass cytometry. Endocrinology. 2024 Sep;165(9): bqae094.
32. Md Moin AS, Dhawan S, Cory M, Butler PC, Rizza RA, Butler AE. Increased frequency of hormone negative and polyhormonal endocrine cells in lean individuals with type 2 diabetes. The Journal of Clinical Endocrinology & Metabolism. 2016 Oct 1;101(10):3628–36.
33. Mezza T, Sorice GP, Conte C, Sun VA, Cefalo CM, Moffa S, et al. β-Cell glucose sensitivity is linked to insulin/glucagon bihormonal cells in nondiabetic humans. The Journal of Clinical Endocrinology & Metabolism. 2016 Feb 1;101(2):470–5.
34. Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of adult pancreatic α-cells to β-cells after extreme β-cell loss. Nature. 2010 Apr 22;464(7292):1149–54.
35. Wang MY, Dean ED, Quittner-Strom E, Zhu Y, Chowdhury KH, Zhang Z, et al. Glucagon blockade restores functional β-cell mass in type 1 diabetic mice and enhances function of human islets. Proceedings of the National Academy of Sciences. 2021 Mar 2;118(9):e2022142118.
36. Singh S, Pavan MK, Barella LF, Telang J, Shree A, Agarwal S, et al. A Temporal Single-Cell Multi-Omics Atlas of Murine Pancreatic Islet Remodeling During Hyperglycaemia Progression. BioRxiv. 2025 Jun 1:2025–05.
37. Rosselot C, Li Y, Wang P, Alvarsson A, Beliard K, Lu G, et al. Harmine and exendin-4 combination therapy safely expands human β cell mass in vivo in a mouse xenograft system. Science Translational Medicine. 2024 Jul 10;16(755):eadg3456.
38. Karakose E, Wang X, Wang P, Carcamo S, Demircioglu D, Lambertini L, et al. Cycling alpha cells in regenerative drug-treated human pancreatic islets may serve as key beta cell progenitors. Cell Reports Medicine. 2024 Dec 17;5(12):101832.
39. Ben-Othman N, Vieira A, Courtney M, Record F, Gjernes E, Avolio F, et al. Long-term GABA administration induces alpha cell-mediated beta-like cell neogenesis. Cell. 2017 Jan 12;168(1):73–85.
40. Liu N, Liu T, Alim N, Zou J, Hu X, Zhang B, et al. 5-HT promotes pancreatic α-to-β cell transdifferentiation. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research. 2025 Jun 1;1872(5):119958.
41. Hill TG, Hill DJ. The importance of intra-islet communication in the function and plasticity of the islets of Langerhans during health and diabetes. International Journal of Molecular Sciences. 2024 Apr 6;25(7):4070.
42. Wei R, Cui X, Feng J, Gu L, Lang S, Wei T, et al. Dapagliflozin promotes beta cell regeneration by inducing pancreatic endocrine cell phenotype conversion in type 2 diabetic mice. Metabolism. 2020 Oct 1; 111:154324.
43. Pardo CA, Massoz L, Dupont MA, Bergemann D, Bourdouxhe J, Lavergne A, et al. A δ-cell subpopulation with a pro-β-cell identity contributes to efficient age-independent recovery in a zebrafish model of diabetes. Elife. 2022 Jan 21;11: e67576.
44. Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, et al. Diabetes recovery by age-dependent conversion of pancreatic δ-cells into insulin producers. Nature. 2014 Oct 23;514(7523):503–7.
45. Gribben C, Lambert C, Messal HA, Hubber EL, Rackham C, Evans I, et al. Ductal Ngn3-expressing progenitors contribute to adult b cell neogenesis in the pancreas. Cell Stem Cell. 2023 Apr 6; 30:498–9.
46. Téllez N, Vilaseca M, Martí Y, Pla A, Montanya E. β-Cell dedifferentiation, reduced duct cell plasticity, and impaired β-cell mass regeneration in middle-aged rats. American Journal of Physiology-Endocrinology and Metabolism. 2016 Sep 1;311(3): E554–63.
47. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. β-cell replication is the primary mechanism subserving the postnatal expansion of β-cell mass in humans. Diabetes. 2008 Jun 1;57(6):1584–94.
48. Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, et al. Significant human β-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. The Journal of Clinical Endocrinology & Metabolism. 2010 Oct 1;95(10): E234–9.
49. Spijker HS, Ravelli RB, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, et al. Conversion of mature human β-cells into glucagon-producing α-cells. Diabetes. 2013 Jul 1;62(7):2471-80.
50. Webster KL, Mirmira RG. Beta cell dedifferentiation in type 1 diabetes: sacrificing function for survival? Frontiers in Endocrinology. 2024 Jun 6; 15:1427723.
51. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metabolism. 2014 Feb 4;19(2):259–71.
52. Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of β-cell dedifferentiation in human type 2 diabetes. The Journal of Clinical Endocrinology & Metabolism. 2016 Mar 1;101(3):1044–54.
53. Kim-Muller JY, Fan J, Kim YJ, Lee SA, Ishida E, Blaner WS, et al. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic β cells in diabetic mice. Nature Communications. 2016 Aug 30;7(1):12631.
54. Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. Identification of genes selectively disallowed in the pancreatic islet. Islets. 2010 Mar 1;2(2):89–95.
55. Stojanovic J, Andjelic-Jelic M, Vuksanovic M, Marjanovic-Petkovic M, Jojic B, Stojanovic M, et al. The effects of early short-term insulin treatment vs. glimepiride on beta cell function in newly diagnosed type 2 diabetes with HbA1c above 9%. Turkish Journal of Medical Sciences. 2023;53(2):552–62.
56. Ishida E, Kim-Muller JY, Accili D. Pair feeding, but not insulin, phloridzin, or rosiglitazone treatment, curtails markers of β-cell dedifferentiation in db/db mice. Diabetes. 2017 Aug 1;66(8):2092–101.
57. Li B, Cheng Y, Yin Y, Xue J, Yu S, Gao J, et al. Reversion of early-and late-stage β-cell dedifferentiation by human umbilical cord-derived mesenchymal stem cells in type 2 diabetic mice. Cytotherapy. 2021 Jun 1;23(6):510–20.