Abstract
Lipids are an essential class of complex biomolecules involved in maintaining cellular energy homeostasis, structural organization and signal transduction. Dysregulated lipid signaling and metabolism have increasingly been reported in various pathological settings like diabetes, cardiomyopathy, neurological pathologies, malignancies and infectious diseases. Recent technological advances in metabolomics and lipidomics have shown enormous complexities and functionalities of lipids. The role of lipid metabolism in maintaining cancer heterogeneity and plasticity is an important hallmark in the disease progression, which is often associated with reduced drug efficacy and poor survival outcomes in patients. In the last decade different research groups have comprehensively shown using variety of cell lines and in vivo tumor models that cancer cells show higher dependency on fats, especially during stresses, like drug insults, oxidative damage, immune attack and metastasis. This metabolic shift helps metastasizing cells to extract ATP and NADPH via β-oxidation to curtail oxidative stress and create an immunosuppressive niche.
Lipids play a crucial role in pathogenesis of various infectious diseases such as HIV, SARS-CoV-2, and influenza, and are indispensable in Mtb survival within the host. In this review, we explore the impact of altered lipidomics and dysregulated signaling in shielding cancer and tuberculosis from various internal and external stresses, as well as their potential as a novel therapeutic target for managing diseases.
Keywords
Cancer, Lipid signaling, Signal transduction, Tuberculosis
Introduction
Lipids play a multifaceted role in cellular physiology and pathology. They primarily consist of fatty acids, triglycerides, sterols (nonglycerides), phospholipids and lipoproteins, which are crucial in regulating diverse cellular functions such as maintenance of cellular energetics, structural integrity, and signaling. Dysregulated lipid metabolism has been associated with various diseases, including cancer and tuberculosis [1-3].
Rewired cancer metabolism is an important hallmark of tumorigenesis [2]. Tumor metabolic heterogeneity is associated with drug resistance, reduced survival and poor patient outcomes [4]. While processes like Warburg effect and glucose metabolism have been extensively studied in cancer and other diseases, recent advancements in metabolomics have highlighted the role of lipids in various pathological settings, particularly in cancer and tuberculosis [5,6]. Lipids serve as a key metabolite for tumor initiation, progression and immune evasion. At various stages of tumor development and drug resistance, cancer cells exhibit enhanced lipid synthesis, storage, and degradation, and disrupting these processes lead to tumor regression and cell death [7].
Lipid metabolism dysregulation is not exclusive to tumor cells only, it also plays a key role in neurological disorders, cardiomyopathy, diabetes and infectious diseases such as tuberculosis [8]. Emerging evidence has shown association between alteration in lipid metabolism and tuberculosis progression (Figure 1), which is often linked to therapy failure and development of multi-drug resistant phenotypes [9].
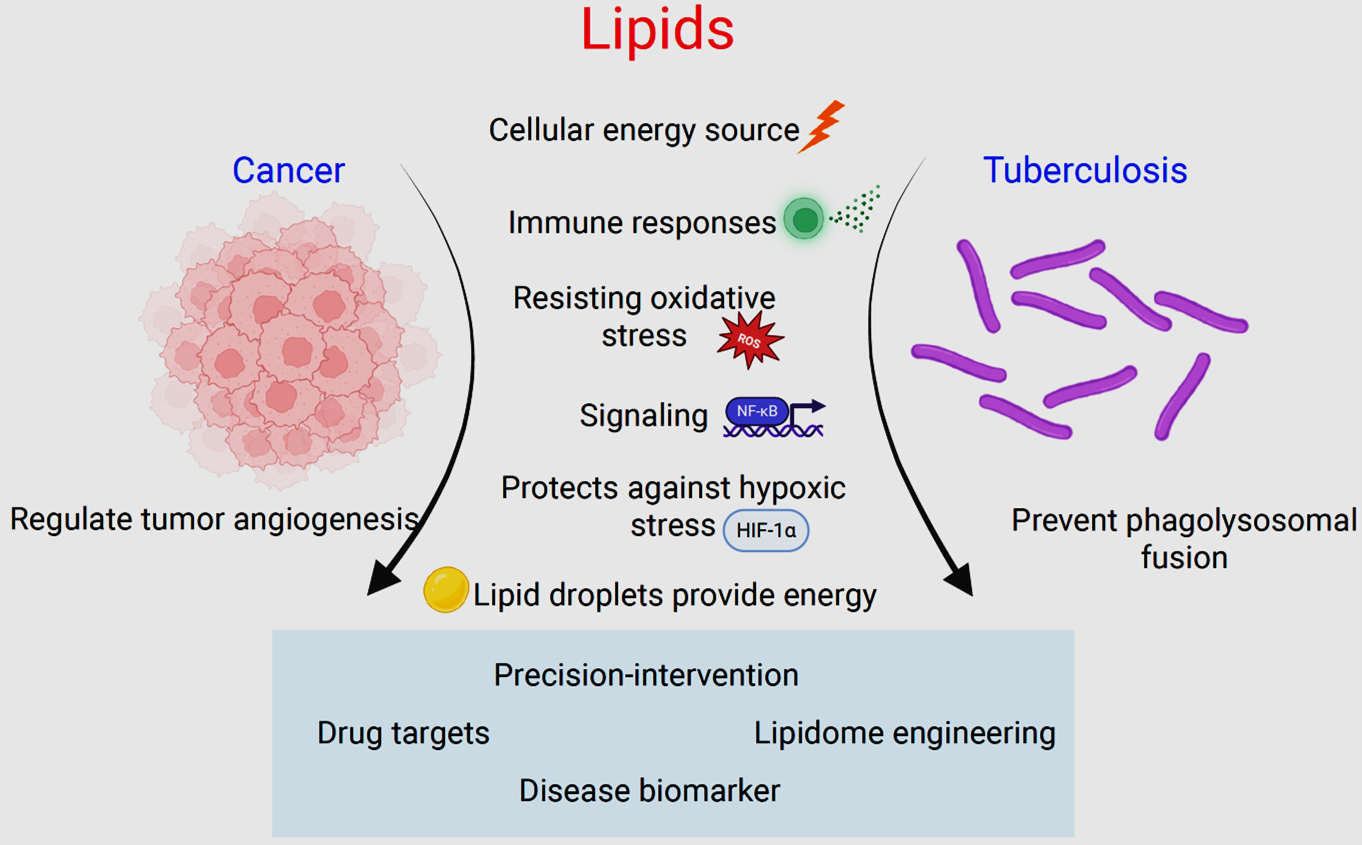
In this report, we summarize recent advancements in lipidomics and its role in cancer and tuberculosis, drawing parallels between these two pathologies and how they exploit lipids in their progression. Finally, we discuss the potential of targeting lipid metabolism in preclinical and clinical settings.
Lipid Signaling in Disease Pathology
Dysregulated signaling is a common phenomenon in various pathologies including cancer. Overactivated signaling involved in lipid metabolism rewiring has been extensively studied in different cancer models. For instance, one of the metabolic features of renal cell clear cell carcinoma (ccRCC) is elevated lipid storage and signaling under hypoxia, which is often associated with poor prognosis in patients [10]. To examine this, Qiu et al. explored the role of HIF2α (hypoxia-inducible factor 2α) in regulating lipid metabolism and showed that HIF2α signaling promotes lipid storage, ER fitness, and survival through PLIN2 upregulation. Furthermore, they found that the inhibition of PLIN2 activity led to decreased lipid storage and reduced survival of renal cancer cells [11]. Similarly, Bensaad et al. showed HIF1α promotes increased uptake of fatty acid and lipid storage in breast cancer and glioblastoma cells and targeting lipid accumulation led to reduced tumor load in mice model [12].
The PI3K/AKT pathway is well known for its role in cellular proliferation and biomass generation in both physiological and pathological settings [13,14]. Many recent studies suggest that its dysregulation contributes to lipid metabolism alterations and reprogramming in cancer. Bengoechea-Alonso et al. showed that AKT mediated inactivation of GSK3 leads to stabilization of SREBP1 and de novo lipid biogenesis, while its inhibition led to cell cycle arrest. Similarly, Li et al. found that CD147-mediated activation of SREBP1c via AKT/mTOR signaling led to the upregulation of de novo lipogenesis genes FASN and ACC in liver cancer [15-17]. Another key oncogene involved in cancer metabolic reprograming and biomass generation is MYC. Aberrant MYC signaling is a well-documented phenomenon in many cancers. In fact, some cancer types are highly addicted to its function due to its role in cellular transformation and growth. Interestingly, it has been observed that MYC transcriptionally regulates important enzymes like FASN and SCD, thus modulating de novo synthesis of lipids. Inhibition of MYC’s activity showed increased lipid droplets accumulation, dysregulated lipid and redox homeostasis and increased cell death [18-20]. An extensive discussion of various other pathways and proteins involved in crucial regulation of lipid signaling, including their interacting partners, mechanism of action, and biological significance, is discussed elsewhere in detail [21].
Importance of lipids in maintaining structural components and energy dynamics
Lipids play an essential role in maintaining cellular structures and regulating energy homeostasis [1]. They form plasma membrane, nuclear membrane, and membranes of all organelles within the cell, providing compartmentalization for spatiotemporal functions and regulatory processes. Lipids enable cells to adapt to fluctuations during changes in cell shape, size, and external stimuli, which are often associated with pathological consequences [22]. Numerous proteins also interact with lipids to provide structural integrity and facilitate cellular signaling [23]. Each major class of lipids is destined for a distinct role: triglycerides mainly function as storehouse for fatty acids routed to mitochondria and peroxisome for energy generation and signaling via diacylglycerol. Phospholipids maintain the membrane polarity and flexibility, while cholesterol is crucial for maintaining fluidity in the lipid bilayer and serves as a structural backbone for various steroid hormone synthesis [23-25].
Notably, when it comes to energy generation, only a few classes of saturated fatty acids and short chain fatty acids undergo degradation and contribute to maintaining cellular energy homeostasis. Cancer cells employ various mechanisms to regulate dynamic lipid metabolism, including enhanced lipid droplet synthesis, transport from tumor microenvironment, enhanced expression of de novo fatty acids synthesis genes, and reliance on neighboring fibroblasts and adipocytes for supply of fat molecules [26-28]. Free fatty acids released by adipocytes become a substrate for β-oxidation in the mitochondria, fueling tumor growth. To explore metabolic flexibility and lipid dependency in hepatocellular carcinoma (HCC) induced by β-catenin oncogene, Senni et al. showed that progression of HCC is dependent on fatty acid oxidation (FAO), rather than glycolysis [29]. Furthermore, inhibiting the activity of PPARα, a transcription factor known for regulating expression of lipid metabolic genes, was sufficient to block the development of HCC in mice. Similarly, Zuagg et al. showed that carnitine palmitoyltransferase 1C (CPT1C) overexpressing cells were resistant towards glucose deprivation and hypoxia, and more dependent on FAO for ATP production. Additionally, knockdown of CPT1C led to apoptosis, inhibited ATP production in cell line and caused regression of tumor xenograft post metformin treatment in mice [30]. In contrast, stimulating fatty acid catabolism in tumor infiltrating CD8+ (TIL) cells has been shown to improve immunotherapy response and reduce tumor burden in melanoma [31].
Lipids as an important regulator of metabolic and oxidative stress during metastasis
Metabolic adaptability is frequently observed in the metastatic cascades of various human tumors [32]. Tumor cells exhibit dynamic instability in lipid metabolism to counter oxidative stress and meet energy demand during epithelial mesenchymal transition (EMT). One of the strategies cancer cells employ to cope with increased oxidative stress while migrating is shifting their metabolic reliance towards β-oxidation of fats. This shift has two key advantages: first, it provides ample energy for anabolic activities; second, it generates a pool of NADPH molecules, which is a crucial cofactor for enzymes responsible for lowering down oxidative stress [33-35].
Fatty acid degradation shields cancer cells from multiple cell death pathways triggered by metabolic stress and matrix detachment [35,36]. Li et al. investigated how cancer cells exploit FAO to overcome metabolic stress and loss of attachment. Using melanoma cell line and mice model they found that glucose deprivation activates ERK2 mediated Nur77 translocation to mitochondria and facilitate FAO to generate NADPH and ATP, which prevented ROS generation [37]. Further, they found Nur77 and TPβ interaction was essential for survival of circulating tumor cells and metastasis. Similarly, Lee et al. found that during lymph node metastasis metabolic dependency towards FAO is essential for growth and survival of migrating cancer cells [38]. To examine the potential of FAO blockade in drug-resistant mesenchymal cancer cells, Li et al. used MDA-MB231 cells and showed that inhibiting FAO in these cells reduces their potential to migrate, invade, and prime them towards apoptosis. This inhibition also led to decreased mitochondrial membrane potential [39]. Similarly, Wright et al. found that UB-domain containing protein 1 (CDCP1) promotes metastasis of triple negative breast cancer cells by leveraging β-oxidation and targeting lipid droplets [40]. Previously, it has been shown by various groups that metastasizing cells, which are resistant to therapy show alterations in lipid synthesis, uptake and breakdown. For instance, Pascual et al. showed that mice bearing facial tumor had higher metastasis initiating cells and enhanced expression of CD36 and lipid metabolism genes when fed with high fat diet compared to low fat fed mice [41,42]. Similar results were obtained in other investigations, wherein they showed high fat diet in colon cancer mice model activated lipid catabolism transcriptional program via PPAR-δ, which in turn lead to increased stemness and liver metastasis from primary tumor [43]. Similarly, Chen et al. showed increased metastatic potential of prostate cancer cells using mice via SREBP mediated upregulation of lipid synthesis [44]. In line with this, Li et al. showed that high fat diet induced obesity promoted lung metastasis in gastric cancer by enhancing the activity of DGAT2 enzyme, which is important for lipid droplet synthesis and neutral fat storage. Interestingly, inhibiting DGAT2 activity, either by shRNA or by pharmacological agents decreased cancer cell’s ability to metastasize and became sensitive to anoikis, a type of programmed cell death occurs due to detachment of cells from extracellular matrix [45].
The role of cholesterol and its metabolites in protecting cancer cells from ferroptosis
Cholesterol is a vital component in the plasma membrane, involved in maintaining lipid raft integrity and signaling. Stochiometric changes in metabolites involved in cholesterol biosynthesis are associated with cancer pathogenesis. Clinical studies have shown that hypercholesterolemia is associated with reduced survival outcomes in cancer patients with advanced disease [46,47]. Recent research has highlighted the role of various cholesterol biosynthesis metabolites in manipulating cell death signals and enhance cellular proliferation. For example, several recent have studies shown that higher RNA expression of HMGCR, an important enzyme for cholesterol synthesis, is associated with poor survival in prostate cancer patients and its inhibition induces cell death in glioblastoma [48-50]. In another study by Liu et al. they showed 27-dihydrocholesterol (27HC), an intermediate metabolite in cholesterol synthesis pathway was associated with increased tumorigenicity and metastasis and enhanced resistance towards ferroptosis. Interestingly, inhibiting GPX4 gene’s (a lipid-repair enzyme) activity sensitized them towards ferroptosis [51].
Notably, the potential of intermediate metabolites from cholesterol synthesis pathway in inhibiting ferroptosis by blocking peroxidation of phospholipids has been highlighted elegantly recently by two different research studies. In the first study, wherein Li et al. used genome wide CRISPR–Cas9 screening approach to fish out factors involved in regulating ferroptosis in HEK293T cells and found several well-known genes regulating this process. Interestingly, they found 7-DHC reductase (DHCR7) as the top hit in this group, which is involved in converting 7-dehydrocholesterol (7-DHC) to cholesterol. Mechanistically, they found that 7-dehydrocholesterol (7-DHC) acts like a free radical quencher due to its conjugated dienes present in the sterol core. This was associated with decreased lipid peroxidation of phospholipids in both the plasma membrane and mitochondria’s lipid bilayer, thereby inhibiting ferroptosis [52]. In the second study by Freitas et al. wherein they took a slightly different approach by using an inducible GPX4 knock out cell line Pfa1, an important gene in protecting lipid damage, to find genes conferring resistance towards ferroptosis using similar (CRISPR-Cas9) genetic screen approach. They also found DHCR7 as one of the top hits under RAS-selective lethal 3 (RSL3) stress, an inhibitor of GPX4. To show its potential in protecting phospholipids’ peroxidation, especially in the plasma membrane, they used iron/ascorbate oxidation model for membrane permeabilization and found that incorporation of liposomes containing 7-DHC enhanced resistance towards ferroptosis. Additionally, using mice model, they showed that deletion of DHCR7 (which enhanced accumulation of 7-DHC) led to increased aggressiveness of neuroblastoma and lymphomas cancer cells [53]. This highlights the role of cholesterol in protecting cancer cells from phospholipid peroxidation and escaping ferroptosis, which is triggered by increased oxidative stress.
Role of lipids in manipulating the immune system in the tumor microenvironment
Cholesterol biosynthetic metabolites not only protect cancer cells from oxidative damage but also contribute to the immune suppression in the tumor microenvironment. In an interesting study led by Baek et al. they showed that 27HC promotes metastasis in breast cancer by acting on myeloid cells and creating an immunosuppressive niche for tumor growth [54]. Furthermore, ablating CYPA271, the rate limiting enzyme in 27HC synthesis led to the decreased metastasis in mice model by manipulating the functions of γδ-T cells and polymorphonuclear-neutrophils. Similarly, Goossens et al. used metastatic ovarian cancer mice model to demonstrate how cholesterol dependent reprogramming supports cancer progression by converting tumoricidal macrophages to tumor promoting ones and lipid rafts mediated IL-4 activation and decreased expression of IFNγ target genes. Additionally, they showed that the ablation of cholesterol efflux transporter genes led to the reversal of tumor-associated macrophage function and a decreased tumor load [55].
Apart from cholesterol, other lipid species are also linked in impeding immune cell functions and decreased tumoricidal effect in the tumor niche by reprogramming lipid metabolism and immune signaling pathways. A study by Park et al. demonstrated that cancer cells dampen immune responses by secreting macrophage colony-stimulating factors (M-CSF), which activate tumor myeloid cells. This in turn activates fatty acid synthase gene and changes nuclear gene expression via Peroxisome Proliferator-Activated Receptor β/δ (PPARβ/δ) [56]. Similarly, Vegila et al. demonstrated that the upregulation of fatty acid transporter gene (FAT2) in polymorphonuclear myeloid-derived suppressor cells (MDSCs) induces immunosuppressive environment and promotes tumorigenesis. Their study revealed that FAT2 expression is regulated by M-CSF through STAT3 signaling and its deletion abrogated the tumor suppressive behavior of MDSCs. Furthermore, they showed that a combined blockade of FAT2 along with immune check-point inhibition regressed tumor growth in mice [57].
Dysfunctional T cell immunity has been observed under metabolic stresses, particularly, in lipid-enriched tumor microenvironment [58]. Xu et al. demonstrated how presence of oxidized lipids can influence T cell activity and shape tumor immunity. Using mice model, they found that the accumulation of various oxidized lipids in the tumor microenvironment led to progressive decline in T cell function. Mechanistically, it was shown that CD36, a scavenger receptor for oxidized lipids facilitated the uptake of oxidized low-density lipoproteins (OxLDL), which increased T cell’s lipid peroxidation and activated p38 signaling. Interestingly, they found reversal of T cell function either by blocking p38 kinase activation or overexpressing glutathione peroxidase 4 (GPX4), a phospholipid hydroperoxides responsible for preventing lipid peroxidation [59].
Role of Lipids in Tuberculosis
Lipids form structural niches during Mtb infection and disease progression
Tuberculosis (TB) is the leading cause of death from an infectious disease, with around 1.25 million deaths per year [60]. Mycobacterium tuberculosis (Mtb), the causative agent of TB, is a pathogenic bacterium that requires specialized genes and systems to evade host immune responses and persist within the host for long periods. Approximately 250 Mtb genes are implicated in lipid uptake, synthesis, degradation, and modification [61,62]. This highlights Mtb's preference for utilizing lipids over carbohydrates [63,64]. The dynamic nature of lipid metabolism at different stages of infection influences disease progression and therapy response. Various signaling pathways and proteins coordinate the process of infection, survival, and evasion of the immune system, and therapy involving Mtb's lipidome [65].
During infection, Mtb breaks down host lipids, shifting its metabolism toward host lipid catabolism to accumulate precursors for carbon and energy sources (lipid anabolism), which supports virulence. Various host and Mtb factors are involved in fostering foamy cell formation because of metabolic calibration. Many lipid derivatives are generated from the degradation of the surrounding foamy cells by necrosis or promoted by IFN-γ. IFN released by T-lymphocytes stimulates nearby macrophages to promote necrotic cell death of foamy macrophages [65,66]. This results in the accumulation of lipids such as cholesterol, cholesteryl esters, triacylglycerols, and lactosylceramides within the caseous foci, creating a nutrient-rich environment for the extracellular bacilli [67]. In addition, caseous granulomas display increased expression of adipophilin (ADFP), which is involved in lipid modulation and is detected on the surface and the core of lipid droplets and in necrotic centers, suggesting that it comes from foamy macrophages [68]. Host lipid gene expression also increases in granulomas. Mtb can utilize sterol rings and side chains of cholesterol even inside IFN-γ-activated macrophages and achieve persistence [69,70].
The PI3K pathway plays a significant role in Mtb infection and immune evasion. Transcriptomic analysis of TB patients has identified a set of molecules linked to the PI3K pathway that are dysregulated in active tuberculosis. Hyperactivation of the PI3K pathway leads to the prolonged life span of neutrophils, increasing inflammation and tissue damage [71]. However, the dysregulated PI3K pathway leads to disruption of phagolysosomal fusion, a crucial step in the immune response against Mtb [72]. PIK3IP1, a protein involved in regulating the PI3K pathway, is downregulated in TB patients, contributing to neutrophil persistence. Targeting PI3K and its regulators may offer host-directed therapeutic strategies for TB treatment. PIK3C2B, another protein involved in the PI3K pathway and expressed in leukocytes, is also downregulated in TB patients [71,72].
Hypoxia-inducible factor 1 alpha (HIF-1α) is a transcription factor that plays a crucial role in Mtb infection and immune evasion [73]. It is activated in the hypoxic environment of TB granulomas and regulates lipid metabolism to adapt to low-oxygen conditions. HIF-1α promotes lipid uptake and storage while suppressing oxidative metabolism. Additionally, HIF-1α drives the expression of genes involved in fatty acid utilization and lipid droplet formation during hypoxia [74]. Mtb induces and promotes foamy cell formation by inducing the HIF-1-mediated glyoxylate pathway and inhibiting the β-oxidation pathway. Several infections use the accumulation of lipid droplets (LDs), which is known to bolster intracellular bacterial persistence. HIF-1α also plays a key role in LD formation, acting through the IFN-γ/HIF-1α/Hig2 signaling axis [75].
Peroxisome proliferator-activated receptor-gamma (PPAR-γ), a member of the PPAR family that regulates lipid and glucose metabolism as well as energy balance, along with testicular receptor 4 (TR4), facilitates Mtb survival within host cells by promoting lipid accumulation in foamy macrophages. Deleting Pparg and Nr2C2 genes, which encode PPAR-γ and TR4, respectively, reduces Mtb's intracellular persistence by limiting lipid uptake through the scavenger receptor CD36, disrupting the lipid-rich environment essential for Mtb survival [61]. Additionally, ketomycolic acid, an oxygenated derivative of mycolic acid in Mtb, can activate TR4, forming foamy macrophages within granulomas. Lowering lipid levels can also lower the growth of intracellular bacteria. PPAR-γ activation contributes to foam cell formation, increasing IL-10 production and enhancing mycobacterial persistence. PPAR-γ supports lipid droplet formation and favors Mtb survival in hepatocytes [76]. On the other hand, PPAR-α induction by Mtb infection increases lipid catabolism, β-oxidation and autophagy in macrophages [77]. PPAR-α agonists could serve as host-directed therapies by limiting mycobacterial access to host lipids while boosting autophagic clearance [77,78].
Mtb also influences nuclear transcription factors signaling pathways to manipulate host metabolism. For instance, macrophage infection triggers TNF receptor signaling, leading to downstream activation of the caspase cascade and mTORC1, which promotes LD accumulation by reducing fatty acid consumption [79].
Cholesterol: the safe haven from infection to latency in Mtb pathogenesis
Cholesterol metabolism is one of the major sources of carbon and is crucial for optimal growth and persistence, particularly during the chronic phase of Mtb infection. Studies have shown that Mtb increases the uptake of low-density lipoprotein (LDL) and shows enhanced de novo cholesterol synthesis in macrophages post-infection. Furthermore, Mtb actively inhibits cholesterol efflux from macrophages via an ATP-binding cassette (ABC) transporter, leading to lipid droplet synthesis and granuloma formation, key events in systemic dissemination [80]. Among the various operons facilitating Mtb invasion and intracellular survival, the mce4 operon, which includes igr genes, plays an essential role in intracellular cholesterol utilization. While igr mutants grow optimally on fatty acids and under in vitro stress, their survival is severely impaired when cholesterol is the primary carbon source, indicating that the igr transporter system is crucial for complete cholesterol metabolism [80,81]. Cholesterol catabolism is an essential source of carbon and a key virulence factor in establishing TB infection. Once inside Mtb, cholesterol and fatty acids serve as crucial metabolic resources, contributing to the synthesis of key lipids such as PDIM), PAT, and mycolic acids, all essential for bacterial growth and survival. β-oxidation generates byproducts that aid Mtb in persisting in chronic infection and dampening immune responses. Even-chain fatty acids are catabolized into acetyl-CoA, while odd-chain fatty acids generate acetyl-CoA and propionyl-CoA [82-86]. Jain et al. showed that the bacterium prevents the toxic accumulation of propionyl CoA by synthesizing methyl malonyl CoA (MMCoA) lipids like polyacyl trehalose, Phthiocerol Dimycocerosate (PDIM), and Sulfolipid-1 (SL-1), which are important players in Mtb virulence [87].
The Mtb genome encodes several genes involved in the cholesterol catabolic process, such as ipdAB, kstR2, and kshAB, critical for maintaining virulence post-infection [88]. Mutants of cholesterol catabolism genes lead to reduced virulence in mice [88,89]. This shows how vital cholesterol is as an energy source during infection, and the inability to transport or utilize cholesterol affects Mtb's ability to escape immune clearance. IpdAB participates in the cholesterol degradation pathway by catalyzing the breakdown of side chains of cholesterol intermediates. Disruption of ipdAB gene leads to accumulation of toxic intermediates, resulting in impaired cholesterol utilization and reduced virulence in mouse models [89]. KstR2 is a transcriptional regulator controlling gene expression in the later stages of cholesterol catabolism. Deleting kstR2 leads to dysregulated cholesterol metabolism and intracellular growth defects [90]. KshAB is a Rieske-type oxygenase system responsible for the cleavage of cholesterol's steroid nucleus. Inactivation of kshAB prevents complete cholesterol degradation, leading to impaired Mtb persistence and attenuation in vivo [91,92]. Mtb thrives under oxidative insults. Prakhar et al. observed that cholesterol is one of the major regulators of the antioxidant gene expression program, which helps mitigate oxidative stress inside the macrophage. Mechanistically, they showed that G9a, along with SREBP2, orchestrated the cholesterol biosynthesis program, and the accumulated cholesterol inside the macrophage led to enhanced expression of oxidative genes and survival of Mtb [93]. Similarly, Jin et al. showed that the cells with high cholesterol build- up upregulate antioxidants such as NRF2 and HO-1, two very important players known to be involved in mitigating oxidative stress [94].
Host lipid metabolism and elevated cholesterol levels significantly influence tuberculosis progression. Martens et al. showed that apolipoprotein E-deficient (ApoE(-/-)) mice developed hypercholesterolemia, which was associated with accelerated susceptibility to Mtb infection [95]. Mice that were fed a high-cholesterol diet died within four weeks with a 100% mortality rate. These mice exhibited severe lung pathologies, including abscess-like lesions, increased granulocyte infiltration, and enhanced bacterial load compared to mice fed a low-cholesterol diet. Moreover, there was a delay in adaptive immune response and poor antigen priming, which was associated with reduced gamma interferon production and reduced control of Mtb growth [95]. This suggests that elevated serum cholesterol affects Mtb survival and disrupts host immunity, creating a permissive environment for tuberculosis progression. It is also known that cholesterol is required for retention of host protein coronin-1 (called P57 or TACO), and this retention in mycobacteria-containing phagosomes played a role in deterring phagolysosome fusion [96,97].
These findings establish a critical link between host cholesterol metabolism and tuberculosis pathogenesis, emphasizing cholesterol's dual role in bacterial survival and immune modulation. Understanding these intricate interactions could pave the way for novel therapeutic targets in tuberculosis management, particularly among cohorts with dysregulated lipid and cholesterol metabolism.
Lipids; the key-drivers behind the emergence of drug-resistant phenotypes in tuberculosis
The bacterial cell wall, at an interface between bacteria and the host stresses, is composed of an assortment of lipids, including mycolic acids, which are the major direct or indirect targets of most of the anti-TB drugs [61]. Tuberculosis is a chronic disease with long treatment periods and hence the development of drug resistance during therapy is not uncommon. One of the factors that is associated with reduced effect of antibiotics and failure in therapy is the dysregulated lipidome [9,87]. Metabolic indicators such as presence of lipid bodies in patient’s sputum is associated with poor clinical outcomes [9]. Recently, an untargeted lipidomics study showed a strong correlation between failure in therapy and dysregulated lipid signatures in tuberculosis management. Cohort with failed treatment exhibited lower cholesteryl ester and higher ceramide, sphingomyelin, and triacylglycerol levels compared to successfully treated patients. This suggests that modulating these lipids and related pathways prior to therapy may enhance the efficacy of antibiotics and therapeutics, potentially reducing treatment failures in TB management [79]. Two of the first-line anti-TB drugs, Isoniazid (INH) and ethambutol (ETH) target mycobacterial cell wall. Selective upregulation of lipid metabolic genes under drug stress is seen in patients undergoing therapy. INH treatment has been shown to induce expression of several genes involved in fatty acid metabolism such as tgs1 (a triacylglycerol synthase), prpC (a methylcitrate synthase), fabG4, fadE5, fadA, fadB, fadE10, fadE23, and fadE24, were upregulated under INH drug treatment [98-100]. Reports also suggest that upregulation of fadE23 and fadE24 after INH treatment might play a role in cholesterol recycling [101]. Rifampicin also alters the expression of lipid-metabolic genes. RIF treatment led to downregulation of fabD (mycolic acid synthesis), fadD10 (lipid degradation), fcoT and nrp (involved in PDIM synthesis) [102,103]. Drugs targeting cholesterol, such as statins, significantly lowered the duration of TB treatment [104]. Similarly, HMG CoA reductase inhibitor atorvastatin treatment reduced cholesterol accumulation in the cell wall and increased drug efficacy [105].
Major association studies of how altered lipid metabolites affect drug treatment outcomes is important to design better drug regimes. Host-directed therapies focus on targeting host cellular processes rather than targeting the pathogen. This approach aims to minimize drug adverse side effects and curb bacterial antibiotic resistance, which is a significant public health challenge.
Lipids create an immune suppressive niche for Mtb survival
The establishment and persistence of Mtb inside various myeloid cell populations determine the disease progression. Mtb possesses a diverse range of proteins and lipid mediators, that influence immunological responses from the host defense system [70,106,107]. Using optically transparent zebrafish model, Cambier et al. showed that Mtb and its close relatives such as Mycobacterium marinum use PDIM to mask PAMPs and inhibit nitrosative stress via Toll-like receptor (TLR) that arise from macrophages [108]. Similarly, Mittal et al. showed how PDIM manipulates autophagosome machinery and impairs B-cell function in mice. Mechanistically, they showed PDIM inhibited LC-3-mediated phagocytosis and NADPH oxidase recruitment, which led to an alteration in B-cell accumulation in lymphoid follicles [109]. Another study by Cambier et al. showed how Mtb piggybacks immune cells and mediators for systemic dissemination [110]. Membrane phenolic glycolipid (PGL) triggered local macrophages' STING cytosolic sensing pathway, which resulted in the chemokine CCL2 production. This, in turn, attracted circulating CCR2+ monocytes to the infection site and facilitated fusion with infected macrophages, which allowed for bacterial translocation and subsequent dissemination. Interestingly, interrupting this transfer and extending Mtb retention in resident macrophages promoted infection clearance [110,111]. Similarly, a subset of Mtb, isolated from Beijing, showed very high lethality in the murine model due to the presence of a phenolic glycolipid, which is a highly bioactive lipid type. It was shown that this lipid diminished the induction of pro-inflammatory cytokines such as interleukin-6, -12 and TNF-α. This inhibited secretion of these cytokines led to increased bacterial burden and illness in mice [112]. Other glycolipids such as lipoarabinomannans (LAM) and lipomannans (LM) are also reported to dampen innate responses from the host by modulating the receptor-binding activities of macrophages and dendritic cells [113]. This highlights the potential of glycolipids as a novel antigen target for therapeutic interventions.
Future of Lipidomics in Diagnosis and Treatment of Tuberculosis
Epidemiologically, dyslipidemia has been linked with almost every major human disease (Figure 2). This highlights its potential as a biomarker. Emerging evidence has shown that a high-fat diet is a risk factor for malignancies and tuberculosis. Appropriate dietary interventions, particularly low-fat and ketogenic diets, have the potential to benefit cancer and TB patients. Exploring cholesterol-lowering drugs (statins) would be another interesting avenue to test as an adjunct therapeutic agent in the clinic, because of their well-established safety profiles and plethoric effects, which include cholesterol-lowering, immunomodulatory, and anti-inflammatory properties, statins may be able to interfere with host lipid pathways that are used by intracellular pathogens such as Mtb as well as tumor cells.
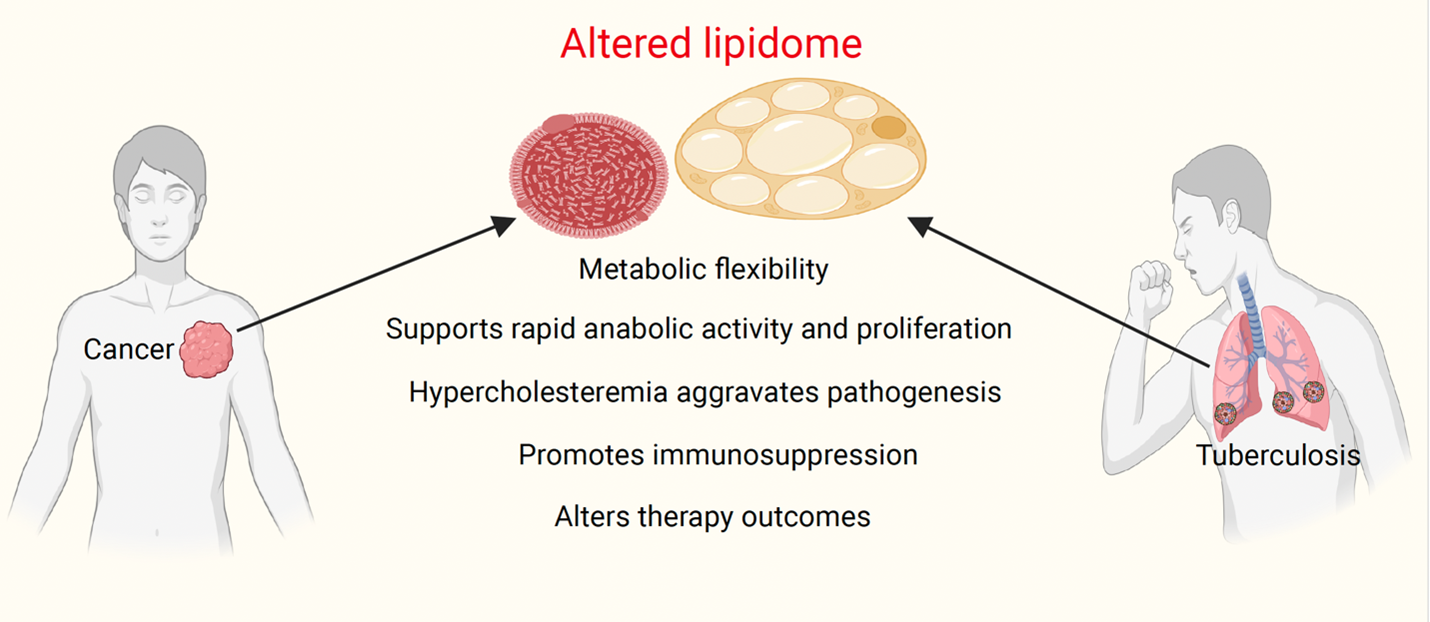
Altered lipidome profiles in TB treatment affect the outcome of TB treatment. In a drug specific manner. However, despite its immense potential, the clinical utility of lipids as a biomarker or therapeutic is yet to be fully achieved. This is partly due to its inherent complexities in biological samples, lack of robust statistical models, and designing of optimized platforms in pre-clinical and clinical settings. For example, in cancer, lipids are utilized in a context-dependent and disease-stage-specific manner. Moreover, cancer cells are highly heterogeneous and can switch their metabolic preferences from fats to glucose under lipid targeting therapies. Also, no single lipid can predict therapy response. Similarly, Mtb shows metabolic flexibility in terms of switching lipid sources under stress. Future success in exploiting lipids as a clinical target will depend on the identification and profiling of context-specific lipid classes in physiology and pathology using advanced biochemical and biophysical techniques, advanced imaging systems, along with the latest artificial intelligence-based approaches. This might help in cataloguing it better and help to establish it as a candidate for various clinical purposes.
References
2. Hanahan D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022 Jan;12(1):31-46.
3. Rombouts Y, Neyrolles O. The fat is in the lysosome: how Mycobacterium tuberculosis tricks macrophages into storing lipids. J Clin Invest. 2023 Mar 15;133(6):e168366.
4. Demicco M, Liu XZ, Leithner K, Fendt SM. Metabolic heterogeneity in cancer. Nat Metab. 2024 Jan;6(1):18-38.
5. DeBerardinis RJ, Chandel NS. We need to talk about the Warburg effect. Nat Metab. 2020 Feb;2(2):127-9.
6. Slayden RA, Barry CE 3rd. Analysis of the Lipids of Mycobacterium tuberculosis. Methods Mol Med. 2001;54:229-45.
7. Martin-Perez M, Urdiroz-Urricelqui U, Bigas C, Benitah SA. The role of lipids in cancer progression and metastasis. Cell Metab. 2022 Nov 1;34(11):1675-99.
8. Hornburg D, Wu S, Moqri M, Zhou X, Contrepois K, Bararpour N, et al. Dynamic lipidome alterations associated with human health, disease and ageing. Nat Metab. 2023 Sep;5(9):1578-94.
9. Shivakoti R, Newman JW, Hanna LE, Queiroz ATL, Borkowski K, Gupte AN, et al. Host lipidome and tuberculosis treatment failure. Eur Respir J. 2022 Jan 6;59(1):2004532.
10. Wang S, Wang K, Yue D, Yang X, Pan X, Kong F, et al. MT1G induces lipid droplet accumulation through modulation of H3K14 trimethylation accelerating clear cell renal cell carcinoma progression. Br J Cancer. 2024 Sep;131(4):641-54.
11. Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, et al. HIF2α-Dependent Lipid Storage Promotes Endoplasmic Reticulum Homeostasis in Clear-Cell Renal Cell Carcinoma. Cancer Discov. 2015 Jun;5(6):652-67.
12. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014 Oct 9;9(1):349-65.
13. Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K Pathway in Human Disease. Cell. 2017 Aug 10;170(4):605-35.
14. Huang J, Chen L, Wu J, Ai D, Zhang JQ, Chen TG, et al. Targeting the PI3K/AKT/mTOR Signaling Pathway in the Treatment of Human Diseases: Current Status, Trends, and Solutions. J Med Chem. 2022 Dec 22;65(24):16033-61.
15. Bengoechea-Alonso MT, Ericsson J. A phosphorylation cascade controls the degradation of active SREBP1. J Biol Chem. 2009 Feb 27;284(9):5885-95.
16. Bengoechea-Alonso MT, Ericsson J. The phosphorylation-dependent regulation of nuclear SREBP1 during mitosis links lipid metabolism and cell growth. Cell Cycle. 2016 Oct 17;15(20):2753-65.
17. Li J, Huang Q, Long X, Zhang J, Huang X, Aa J, et al. CD147 reprograms fatty acid metabolism in hepatocellular carcinoma cells through Akt/mTOR/SREBP1c and P38/PPARα pathways. J Hepatol. 2015 Dec;63(6):1378-89.
18. Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013 Aug 1;3(8):a014217.
19. Zirath H, Frenzel A, Oliynyk G, Segerström L, Westermark UK, Larsson K, et al. MYC inhibition induces metabolic changes leading to accumulation of lipid droplets in tumor cells. Proc Natl Acad Sci U S A. 2013 Jun 18;110(25):10258-63.
20. Wang H, Lu J, Edmunds LR, Kulkarni S, Dolezal J, Tao J, et al. Coordinated Activities of Multiple Myc-dependent and Myc-independent Biosynthetic Pathways in Hepatoblastoma. J Biol Chem. 2016 Dec 16;291(51):26241-51.
21. Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008 Feb;9(2):162-76.
22. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. 2008 Feb;9(2):112-24.
23. Cook GM. Glycoproteins in membranes. Biol Rev Camb Philos Soc. 1968 Aug;43(3):363-91.
24. Baenke F, Peck B, Miess H, Schulze A. Hooked on fat: the role of lipid synthesis in cancer metabolism and tumour development. Dis Model Mech. 2013 Nov;6(6):1353-63.
25. Raffy S, Teissié J. Control of lipid membrane stability by cholesterol content. Biophys J. 1999 Apr;76(4):2072-80.
26. Ammendolia DA, Bement WM, Brumell JH. Plasma membrane integrity: implications for health and disease. BMC Biol. 2021 Apr 13;19(1):71.
27. Wang X, Zhang C, Bao N. Molecular mechanism of palmitic acid and its derivatives in tumor progression. Front Oncol. 2023 Aug 9;13:1224125.
28. Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019 Mar;20(3):137-55.
29. Senni N, Savall M, Cabrerizo Granados D, Alves-Guerra MC, Sartor C, Lagoutte I, et al. β-catenin-activated hepatocellular carcinomas are addicted to fatty acids. Gut. 2019 Feb;68(2):322-34.
30. Zaugg K, Yao Y, Reilly PT, Kannan K, Kiarash R, Mason J, et al. Carnitine palmitoyltransferase 1C promotes cell survival and tumor growth under conditions of metabolic stress. Genes Dev. 2011 May 15;25(10):1041-51.
31. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell. 2017 Sep 11;32(3):377-91.e9.
32. Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016 Jun 30;166(1):21-45.
33. Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer. 2021 Mar;21(3):162-80.
34. Manley SJ, Liu W, Welch DR. The KISS1 metastasis suppressor appears to reverse the Warburg effect by shifting from glycolysis to mitochondrial beta-oxidation. J Mol Med (Berl). 2017 Sep;95(9):951-63.
35. Broadfield LA, Pane AA, Talebi A, Swinnen JV, Fendt SM. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev Cell. 2021 May 17;56(10):1363-93.
36. Mason JA, Hagel KR, Hawk MA, Schafer ZT. Metabolism during ECM Detachment: Achilles Heel of Cancer Cells? Trends Cancer. 2017 Jul;3(7):475-81.
37. Li XX, Wang ZJ, Zheng Y, Guan YF, Yang PB, Chen X, et al. Nuclear Receptor Nur77 Facilitates Melanoma Cell Survival under Metabolic Stress by Protecting Fatty Acid Oxidation. Mol Cell. 2018 Feb 1;69(3):480-92.e7.
38. Lee CK, Jeong SH, Jang C, Bae H, Kim YH, Park I, et al. Tumor metastasis to lymph nodes requires YAP-dependent metabolic adaptation. Science. 2019 Feb 8;363(6427):644-9.
39. Li YJ, Fahrmann JF, Aftabizadeh M, Zhao Q, Tripathi SC, Zhang C, et al. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 2022 May 31;39(9):110870.
40. Wright HJ, Hou J, Xu B, Cortez M, Potma EO, Tromberg BJ, et al. CDCP1 drives triple-negative breast cancer metastasis through reduction of lipid-droplet abundance and stimulation of fatty acid oxidation. Proc Natl Acad Sci U S A. 2017 Aug 8;114(32):E6556-65.
41. Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017 Jan 5;541(7635):41-5.
42. Pascual G, Domínguez D, Elosúa-Bayes M, Beckedorff F, Laudanna C, Bigas C, et al. Dietary palmitic acid promotes a prometastatic memory via Schwann cells. Nature. 2021 Nov;599(7885):485-90.
43. Wang D, Fu L, Wei J, Xiong Y, DuBois RN. PPARδ Mediates the Effect of Dietary Fat in Promoting Colorectal Cancer Metastasis. Cancer Res. 2019 Sep 1;79(17):4480-90.
44. Chen M, Zhang J, Sampieri K, Clohessy JG, Mendez L, Gonzalez-Billalabeitia E, et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat Genet. 2018 Feb;50(2):206-18.
45. Li S, Wu T, Lu YX, Wang JX, Yu FH, Yang MZ, et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020 Sep;36:101596.
46. Ding X, Zhang W, Li S, Yang H. The role of cholesterol metabolism in cancer. Am J Cancer Res. 2019 Feb 1;9(2):219-27.
47. Murai T. Cholesterol lowering: role in cancer prevention and treatment. Biol Chem. 2015 Jan;396(1):1-11.
48. Zhang C, Zhu N, Li H, Gong Y, Gu J, Shi Y, et al. New dawn for cancer cell death: Emerging role of lipid metabolism. Mol Metab. 2022 Sep;63:101529.
49. Wang X, Huang Z, Wu Q, Prager BC, Mack SC, Yang K, et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017 Sep 15;77(18):4947-60.
50. Longo J, Mullen PJ, Yu R, van Leeuwen JE, Masoomian M, Woon DTS, et al. An actionable sterol-regulated feedback loop modulates statin sensitivity in prostate cancer. Mol Metab. 2019 Jul;25:119-30.
51. Liu W, Chakraborty B, Safi R, Kazmin D, Chang CY, McDonnell DP. Dysregulated cholesterol homeostasis results in resistance to ferroptosis increasing tumorigenicity and metastasis in cancer. Nat Commun. 2021 Aug 24;12(1):5103.
52. Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L, et al. 7-Dehydrocholesterol dictates ferroptosis sensitivity. Nature. 2024 Feb;626(7998):411-8.
53. Freitas FP, Alborzinia H, Dos Santos AF, Nepachalovich P, Pedrera L, Zilka O, et al. 7-Dehydrocholesterol is an endogenous suppressor of ferroptosis. Nature. 2024 Feb;626(7998):401-10.
54. Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. 2017 Oct 11;8(1):864.
55. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V, et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019 Jun 4;29(6):1376-89.e4.
56. Park J, Lee SE, Hur J, Hong EB, Choi JI, Yang JM, et al. M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARβ/δ Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep. 2015 Mar 10;10(9):1614-25.
57. Veglia F, Tyurin VA, Blasi M, De Leo A, Kossenkov AV, Donthireddy L, et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature. 2019 May;569(7754):73-8.
58. Park J, Hsueh PC, Li Z, Ho PC. Microenvironment-driven metabolic adaptations guiding CD8+ T cell anti-tumor immunity. Immunity. 2023 Jan 10;56(1):32-42.
59. Xu S, Chaudhary O, Rodríguez-Morales P, Sun X, Chen D, Zappasodi R, et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8+ T cells in tumors. Immunity. 2021 Jul 13;54(7):1561-77.e7.
60. World Health Organization. Global tuberculosis report 2024. Geneva: World Health Organization. 2024.
61. Kim H, Shin SJ. Revolutionizing control strategies against Mycobacterium tuberculosis infection through selected targeting of lipid metabolism. Cell Mol Life Sci. 2023 Sep 14;80(10):291.
62. Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998 Jun 11;393(6685):537-44.
63. Pisu D, Huang L, Grenier JK, Russell DG. Dual RNA-Seq of Mtb-Infected Macrophages In Vivo Reveals Ontologically Distinct Host-Pathogen Interactions. Cell Rep. 2020 Jan 14;30(2):335-50.e4.
64. Laval T, Chaumont L, Demangel C. Not too fat to fight: The emerging role of macrophage fatty acid metabolism in immunity to Mycobacterium tuberculosis. Immunol Rev. 2021 May;301(1):84-97.
65. van der Klugt T, van den Biggelaar RHGA, Saris A. Host and bacterial lipid metabolism during tuberculosis infections: possibilities to synergise host- and bacteria-directed therapies. Crit Rev Microbiol. 2025 May;51(3):463-83.
66. Kinsella RJ, Fitzpatrick DA, Creevey CJ, McInerney JO. Fatty acid biosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptive evolution, and gene duplication. Proc Natl Acad Sci U S A. 2003 Sep 2;100(18):10320-5.
67. Lee J, Kornfeld H. Interferon-γ Regulates the Death of M. tuberculosis-Infected Macrophages. J Cell Death. 2010 Mar 3;3:1-11.
68. Larigauderie G, Cuaz-Pérolin C, Younes AB, Furman C, Lasselin C, Copin C, et al. Adipophilin increases triglyceride storage in human macrophages by stimulation of biosynthesis and inhibition of beta-oxidation. FEBS J. 2006 Aug;273(15):3498-510.
69. Queiroz A, Riley LW. Bacterial immunostat: Mycobacterium tuberculosis lipids and their role in the host immune response. Rev Soc Bras Med Trop. 2017 Jan-Feb;50(1):9-18.
70. Li J, Chai QY, Zhang Y, Li BX, Wang J, Qiu XB, et al. Mycobacterium tuberculosis Mce3E suppresses host innate immune responses by targeting ERK1/2 signaling. J Immunol. 2015 Apr 15;194(8):3756-67.
71. Leisching GR. Susceptibility to Tuberculosis Is Associated With PI3K-Dependent Increased Mobilization of Neutrophils. Front Immunol. 2018 Jul 17;9:1669.
72. Zhang X, Huang T, Wu Y, Peng W, Xie H, Pan M, et al. Inhibition of the PI3K-Akt-mTOR signaling pathway in T lymphocytes in patients with active tuberculosis. Int J Infect Dis. 2017 Jun;59:110-7.
73. Cimmino F, Avitabile M, Lasorsa VA, Montella A, Pezone L, Cantalupo S, et al. HIF-1 transcription activity: HIF1A driven response in normoxia and in hypoxia. BMC Med Genet. 2019 Feb 26;20(1):37.
74. Knight M, Braverman J, Asfaha K, Gronert K, Stanley S. Lipid droplet formation in Mycobacterium tuberculosis infected macrophages requires IFN-γ/HIF-1α signaling and supports host defense. PLoS Pathog. 2018 Jan 25;14(1):e1006874.
75. Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. HIF-1α Is an Essential Mediator of IFN-γ-Dependent Immunity to Mycobacterium tuberculosis. J Immunol. 2016 Aug 15;197(4):1287-97.
76. Sarkar B, Singh J, Yadav M, Sharma P, Sharma RD, Singh S, et al. PPARγ mediated enhanced lipid biogenesis fuels Mycobacterium tuberculosis growth in a drug-tolerant hepatocyte environment. eLife [Preprint]. 2025. Available from: https://doi.org/10.7554/eLife.103817.1.
77. Kim YS, Lee HM, Kim JK, Yang CS, Kim TS, Jung M, et al. PPAR-α Activation Mediates Innate Host Defense through Induction of TFEB and Lipid Catabolism. J Immunol. 2017 Apr 15;198(8):3283-95.
78. Penas F, Mirkin GA, Vera M, Cevey Á, González CD, Gómez MI, et al. Treatment in vitro with PPARα and PPARγ ligands drives M1-to-M2 polarization of macrophages from T. cruzi-infected mice. Biochim Biophys Acta. 2015 May;1852(5):893-904.
79. Mekonnen D, Derbie A, Mihret A, Yimer SA, Tønjum T, Gelaw B, et al. Lipid droplets and the transcriptome of Mycobacterium tuberculosis from direct sputa: a literature review. Lipids Health Dis. 2021 Oct 3;20(1):129.
80. Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008 Mar 18;105(11):4376-80.
81. Chang JC, Miner MD, Pandey AK, Gill WP, Harik NS, Sassetti CM, et al. igr Genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009 Aug;191(16):5232-9.
82. Thomas ST, VanderVen BC, Sherman DR, Russell DG, Sampson NS. Pathway profiling in Mycobacterium tuberculosis: elucidation of cholesterol-derived catabolite and enzymes that catalyze its metabolism. J Biol Chem. 2011 Dec 23;286(51):43668-78.
83. Thomas ST, Sampson NS. Mycobacterium tuberculosis utilizes a unique heterotetrameric structure for dehydrogenation of the cholesterol side chain. Biochemistry. 2013 Apr 30;52(17):2895-904.
84. Yang M, Guja KE, Thomas ST, Garcia-Diaz M, Sampson NS. A distinct MaoC-like enoyl-CoA hydratase architecture mediates cholesterol catabolism in Mycobacterium tuberculosis. ACS Chem Biol. 2014 Nov 21;9(11):2632-45.
85. Carere J, McKenna SE, Kimber MS, Seah SY. Characterization of an aldolase-dehydrogenase complex from the cholesterol degradation pathway of Mycobacterium tuberculosis. Biochemistry. 2013 May 21;52(20):3502-11.
86. Crowe AM, Casabon I, Brown KL, Liu J, Lian J, Rogalski JC, et al. Catabolism of the Last Two Steroid Rings in Mycobacterium tuberculosis and Other Bacteria. mBio. 2017 Apr 4;8(2):e00321-17.
87. Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007 Mar 20;104(12):5133-8.
88. Griffin JE, Pandey AK, Gilmore SA, Mizrahi V, McKinney JD, Bertozzi CR, et al. Cholesterol catabolism by Mycobacterium tuberculosis requires transcriptional and metabolic adaptations. Chem Biol. 2012 Feb 24;19(2):218-27.
89. Crowe AM, Workman SD, Watanabe N, Worrall LJ, Strynadka NCJ, Eltis LD. IpdAB, a virulence factor in Mycobacterium tuberculosis, is a cholesterol ring-cleaving hydrolase. Proc Natl Acad Sci U S A. 2018 Apr 10;115(15):E3378-87.
90. Casabon I, Zhu SH, Otani H, Liu J, Mohn WW, Eltis LD. Regulation of the KstR2 regulon of Mycobacterium tuberculosis by a cholesterol catabolite. Mol Microbiol. 2013 Sep;89(6):1201-12.
91. Hu Y, van der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, et al. 3-Ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2010 Jan;75(1):107-21.
92. Capyk JK, Casabon I, Gruninger R, Strynadka NC, Eltis LD. Activity of 3-ketosteroid 9α-hydroxylase (KshAB) indicates cholesterol side chain and ring degradation occur simultaneously in Mycobacterium tuberculosis. J Biol Chem. 2011 Nov 25;286(47):40717-24.
93. Prakhar P, Bhatt B, Lohia GK, Shah A, Mukherjee T, Kolthur-Seetharam U, et al. G9a and Sirtuin6 epigenetically modulate host cholesterol accumulation to facilitate mycobacterial survival. PLoS Pathog. 2023 Oct 23;19(10):e1011731.
94. Jin X, Xu Z, Fan R, Wang C, Ji W, Ma Y, et al. HO‑1 alleviates cholesterol‑induced oxidative stress through activation of Nrf2/ERK and inhibition of PI3K/AKT pathways in endothelial cells. Mol Med Rep. 2017 Sep;16(3):3519-27.
95. Martens GW, Arikan MC, Lee J, Ren F, Vallerskog T, Kornfeld H. Hypercholesterolemia impairs immunity to tuberculosis. Infect Immun. 2008 Aug;76(8):3464-72.
96. Deghmane AE, Soualhine H, Bach H, Sendide K, Itoh S, Tam A, et al. Lipoamide dehydrogenase mediates retention of coronin-1 on BCG vacuoles, leading to arrest in phagosome maturation. J Cell Sci. 2007 Aug 15;120(Pt 16):2796-806.
97. BoseDasgupta S, Pieters J. Striking the Right Balance Determines TB or Not TB. Front Immunol. 2014 Oct 8;5:455.
98. Tudó G, Laing K, Mitchison DA, Butcher PD, Waddell SJ. Examining the basis of isoniazid tolerance in nonreplicating Mycobacterium tuberculosis using transcriptional profiling. Future Med Chem. 2010 Aug;2(8):1371-83.
99. Boshoff HI, Myers TG, Copp BR, McNeil MR, Wilson MA, Barry CE 3rd. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. J Biol Chem. 2004 Sep 17;279(38):40174-84.
100. Waddell SJ, Stabler RA, Laing K, Kremer L, Reynolds RC, Besra GS. The use of microarray analysis to determine the gene expression profiles of Mycobacterium tuberculosis in response to anti-bacterial compounds. Tuberculosis (Edinb). 2004;84(3-4):263-74.
101. Wilson M, DeRisi J, Kristensen HH, Imboden P, Rane S, Brown PO, et al. Exploring drug-induced alterations in gene expression in Mycobacterium tuberculosis by microarray hybridization. Proc Natl Acad Sci U S A. 1999 Oct 26;96(22):12833-8.
102. Meneguello JE, Arita GS, Silva JVO, Ghiraldi-Lopes LD, Caleffi-Ferracioli KR, Siqueira VLD, et al. Insight about cell wall remodulation triggered by rifampicin in Mycobacterium tuberculosis. Tuberculosis (Edinb). 2020 Jan;120:101903.
103. de Knegt GJ, Bruning O, ten Kate MT, de Jong M, van Belkum A, Endtz HP, et al. Rifampicin-induced transcriptome response in rifampicin-resistant Mycobacterium tuberculosis. Tuberculosis (Edinb). 2013 Jan;93(1):96-101.
104. Dutta NK, Bruiners N, Pinn ML, Zimmerman MD, Prideaux B, Dartois V, et al. Statin adjunctive therapy shortens the duration of TB treatment in mice. J Antimicrob Chemother. 2016 Jun;71(6):1570-7.
105. Davuluri KS, Singh AK, Singh AV, Chaudhary P, Raman SK, Kushwaha S, et al. Atorvastatin Potentially Reduces Mycobacterial Severity through Its Action on Lipoarabinomannan and Drug Permeability in Granulomas. Microbiol Spectr. 2023 Jan 31;11(2):e0319722.
106. Wang J, Li BX, Ge PP, Li J, Wang Q, Gao GF, et al. Mycobacterium tuberculosis suppresses innate immunity by coopting the host ubiquitin system. Nat Immunol. 2015 Mar;16(3):237-45.
107. Chandra P, Grigsby SJ, Philips JA. Immune evasion and provocation by Mycobacterium tuberculosis. Nat Rev Microbiol. 2022 Dec;20(12):750-66.
108. Cambier CJ, Takaki KK, Larson RP, Hernandez RE, Tobin DM, Urdahl KB, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014 Jan 9;505(7482):218-22.
109. Mittal E, Philips JA. The Mycobacterium tuberculosis lipid, PDIM, inhibits the NADPH oxidase and autophagy. Autophagy. 2025 Mar;21(3):684-5.
110. Cambier CJ, O'Leary SM, O'Sullivan MP, Keane J, Ramakrishnan L. Phenolic Glycolipid Facilitates Mycobacterial Escape from Microbicidal Tissue-Resident Macrophages. Immunity. 2017 Sep 19;47(3):552-65.e4.
111. Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014 Dec 18;159(7):1497-509.
112. Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004 Sep 2;431(7004):84-7.
113. Józefowski S, Sobota A, Kwiatkowska K. How Mycobacterium tuberculosis subverts host immune responses. Bioessays. 2008 Oct;30(10):943-54.