Abstract
Sphingolipids are well-recognized as major players in the pathogenesis of many human diseases, including chronic kidney disease. The kidney is a very sensitive organ to alterations in sphingolipid metabolism. The critical issues to be addressed in this review relate to the role of sphingolipids and enzymes involved in sphingolipid metabolism in the pathogenesis of glomerular diseases with a special focus on podocytes, a key cellular component of the glomerular filtration barrier. Among several sphingolipids, we will highlight the role of ceramide, sphingosine, sphingosine-1-phosphate and ceramide-1-phosphate. Additionally, we will summarize the current knowledge with regard to the use of sphingolipids as therapeutic agents for the treatment of podocyte injury in kidney disease.
Keywords
Kidney, DKD, FSGS, Podocyte, Sphingolipids, SMPDL3b, S1P, C1P, Ceramide
Highlights
• Sphingolipids and related enzymes modulate podocyte function in both DKD and FSGS.
• Ceramide, sphingosine, ceramide-1-phosphate and sphingosine-1-phosphate may directly contribute to the pathogenesis of glomerular diseases.
• Sphingomyelin phosphodiesterase acid-like 3b (SMPDL3b) is a novel enzyme involved in the development of podocyte injury in DKD and FSGS.
• Therapeutic strategies that target cellular sphingolipids might be protective in DKD, FSGS, and more broadly chronic kidney diseases.
Introduction
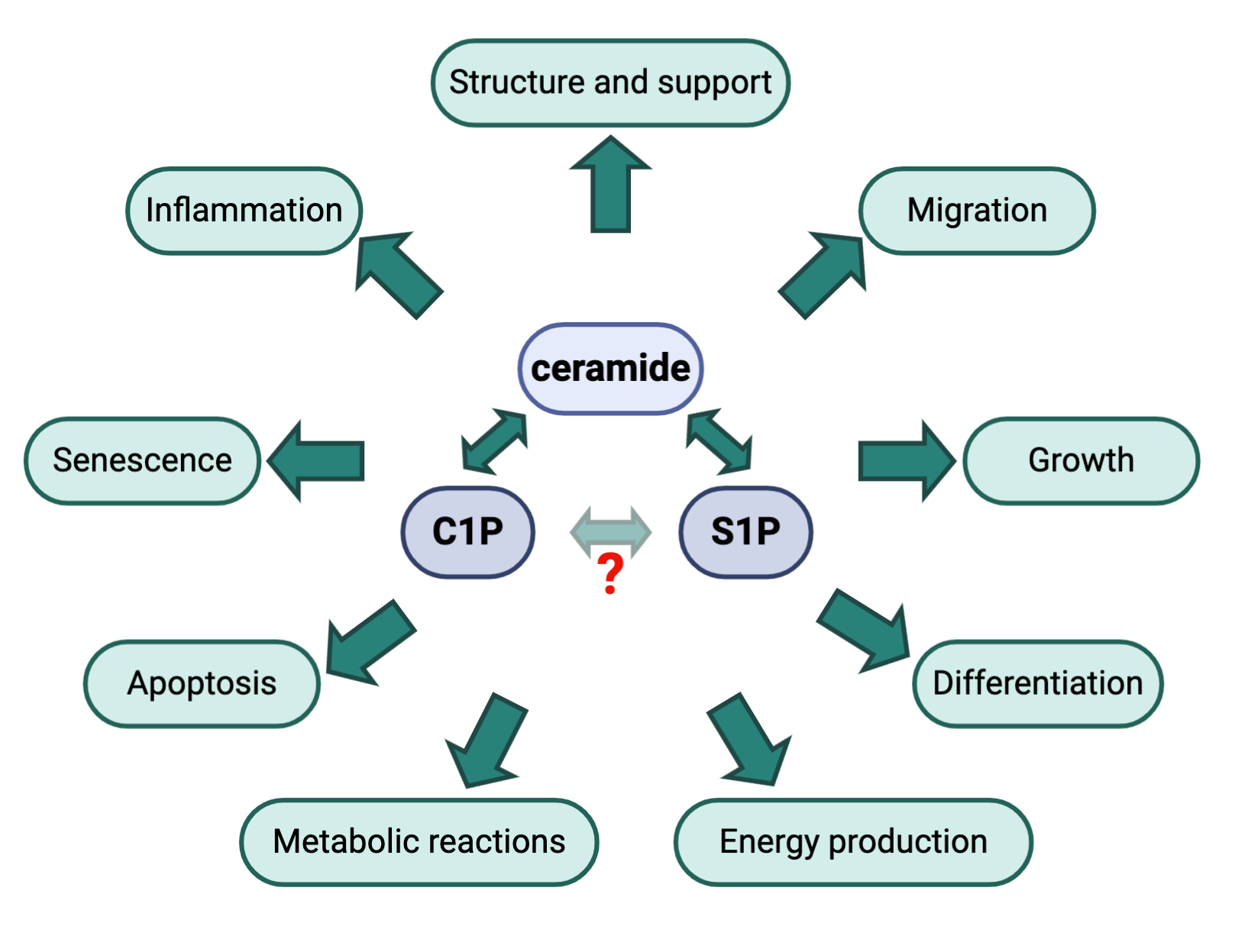
Being a sophisticated and highly organized living system, mammals harbor a large number of biomolecular machineries which represent a dynamic and complex network of interconnections responsible for the effective operation, development and survivability of their body cells. Sphingolipids are a special class of lipids in eukaryotic cells, which have recently gained the attention of researchers because of their involvement in several fundamental processes of living cells, including proliferation and cell death (Figure 1).
Within the kidney, the proper filtration function relies primarily on podocytes, which are terminally differentiated epithelial cells lining the urinary surface of the glomerular capillary tuft. Changes in the function and number of podocytes can lead to the development and progression of glomerular diseases, including diabetic kidney disease (DKD) and focal segmental glomerulosclerosis (FSGS), both of which are common causes of end-stage kidney disease (ESKD) in the USA [1]. However, the cause of podocyte injury and detachment in DKD and FSGS remains largely unknown. Proper filtration function of the glomerular filtration barrier depends heavily on the integrity of lipid raft domains, special regions of the plasma membrane which are enriched in cholesterol, sphingomyelin and glycosyl-phosphatidylinositol (GPI)- anchored proteins. However, renal accumulation of lipid droplets observed both in clinical and experimental glomerular disorders has been shown to correlate with the development of glomerulosclerosis [2-4]. Additionally, seminal studies in experimental DKD established a clear role of glycosphingolipids in its pathogenesis [5]. More recently, podocyte malfunction due to altered metabolism of ceramide-1-phosphate [6, 7] and sphingosine-1-phosphate [8-11] has been described to directly contribute to podocyte injury. In this review we will discuss the contribution of sphingolipids to the pathogenesis of glomerular diseases with a major focus on DKD and FSGS.
Glomerular Diseases: Focus on DKD and FSGS
Chronic kidney disease (CKD) is a condition of gradual loss of kidney function. In 2019, it was estimated that 37 million people, or 15% of US adults, are affected by CKD [12]. Major CKD risk factors include diabetes and high blood pressure, while obesity, heart disease, family history of CKD, and older age may also contribute significantly to kidney damage. Current treatment strategies may slow the decline in kidney function and delay kidney failure, but do not prevent CKD progression. Thus, the development of new treatment options is critical to cure CKD.
Diabetic kidney disease (DKD)
DKD is a major cause of chronic kidney disease leading to ESKD [1]. Clinically, one of the early features of DKD is loss of podocytes. Podocyte loss is an independent predictor of DKD progression in patients with type 1 (T1D) and type 2 (T2D) diabetes [13,14], resulting in a compromised filtration barrier and leakage of proteins into the urine (proteinuria). Several stimuli, such as hyperglycemia, chronic inflammation, oxidative stress and hemodynamic changes have been shown to contribute to podocyte injury in DKD. More recently, altered renal metabolism of lipids such as cholesterol, triglycerides [2,15-17], and sphingolipids has been shown to negatively impact podocyte function and lead to DKD progression and will be reviewed here.
Focal and segmental glomerulosclerosis (FSGS)
FSGS is a histologic lesion that manifests clinically with nephrotic syndrome and is characterized by the presence of sclerotic lesions that affect some glomeruli (focal) and only some area of each glomerulus (segmental). FSGS is the most common cause of primary glomerular disease in adults in the United States and accounts for 10-15% of nephrotic syndrome cases [18]. Irrespective of the form of FSGS, whether primary (idiopathic), secondary, or genetic, loss of podocytes is an important determinant of progression to ESKD [19]. Among different causes of FSGS, recently described familial forms of FSGS demonstrate a causative link between altered sphingolipid metabolism and FSGS [8,9].
Sphingolipid Signaling in Glomerular Disease
Overview of sphingolipid metabolism: focus on the most important metabolites
Sphingolipids were long thought to be passive barrier lipids in cell membranes. Only in the last two decades sphingolipids and their metabolites, the most important being ceramide, sphingosine, ceramide-1-phosphate, and sphingosine-1-phosphate, have been considered as bioactive signaling molecules.
Sphingolipids belong to a large and diverse class of lipids that share a sphingosine base backbone, which is linked to a fatty acid (usually palmitic and stearic acid) via an amide bond, and are known to regulate cell membrane fluidity and structure [20]. The most critical role sphingolipids play in lipid rafts or raft-related caveolae, which are sphingomyelin- and cholesterol-rich microdomains of the plasma membrane, is to regulate protein-protein interactions and intracellular signal transduction. Thus, ceramide accumulation at the plasma membrane results in changes to the biophysical properties of the cell membrane, leading to altered lipid raft composition and changes in signaling properties (as reviewed in Ref. [21]).
Ceramide (Cer) is the central lipid intermediate of sphingolipid metabolism and regulates many of the functions in a cell, particularly anti-proliferative responses, such as growth arrest and/or senescence, and apoptosis (Figure 1). Additionally, ceramides are recognized as tumor-suppressive lipids and the inhibition of ceramidases has proven effective in anticancer therapy [22]. Ceramides can be produced by de novo synthesis from sphingosine (on the surface of the endoplasmic reticulum through the condensation of L-serine and palmitoyl-CoA by serine palmitoyl transferase) by the salvage pathway or from the hydrolysis of sphingomyelin (SM) or other complex sphingolipids by sphingomyelinases [23].
Ceramide kinase (CERK) catalyzes the formation of another bioactive lipid, ceramide-1-phosphate (C1P), from ceramide. The first biological role of C1P was reported in 1995 by Gomez-Munoz et al., who established a role for C1P as a regulator of cellular proliferation and growth [24]. Later on, C1P was described as an anti-apoptotic lipid [25] and, currently, it is well recognized as an important regulator of inflammation (reviewed in Ref. [26]). C1P is also involved in the production of pro-inflammatory eicosanoids, recruiting enzymes responsible for the release of the eicosanoid precursor arachidonate to the plasma membrane [27]. Interestingly, a hypothetical possibility for the direct conversion of C1P to S1P and vice versa has also been suggested [28].
Sphingosine (Sph) is generated from ceramide by the action of ceramidases, primarily alkaline ceramidase 1 (ACER1), and is subsequently converted to sphingosine- 1-phosphate (S1P) by the action of sphingosine kinases (type 1 and type 2). S1P is implicated in cellular growth, survival, migration, angiogenesis and immune reactions (Figure 1). S1P can be dephosphorylated to sphingosine by S1P phosphatase or irreversibly degraded by S1P lyase leading to the formation of ethanolamine-1-phosphate and C16 fatty aldehyde. Interestingly, in the kidney, S1P lyase has been shown to play a role in development of proteinuria in mice [10], while genetic mutations in the gene coding for S1P lyase are associated with severe podocyte injury and nephrotic syndrome in humans [8,9]. Another interesting and important peculiarity of S1P is that it operates both intra-and extra-cellularly, i.e. as an autocrine and paracrine agent [29]. As an autocrine agent, S1P acts through five specific G protein-coupled receptors (designated S1P1-S1P5) and triggers distinctive signaling pathways and cellular responses, some of which can be antagonistic. S1P is primarily produced by erythrocytes and its concentration in blood and tissue may represent a biomarker for some diseases [30-34].
Over the last decades, little attention has been paid to the role of sphingolipids in the kidney and, to their role in podocyte function in health and disease. However, more recently, researchers have actively investigated the role of sphingolipids in the kidney, particularly in glomerular diseases. The accumulation of sphingolipids in glomerular diseases may be of genetic or non-genetic origin. For more information on the role of sphingolipids in glomerular diseases of genetic origin, readers are referred to a review previously published by us [35].
Sphingolipids in DKD
Historically, increased levels of glycosphingolipids [36], ceramide [37], sphingosine and sphinganine [38] have been reported in the plasma of patients with T1D and T2D.
A recent study performed on diabetic C57BLKS db/ db mice demonstrated elevated long-chain ceramides (C14:0, C16:0, C18:0, C20:0) and C18:0 glucosylceramide in plasma, while long-chain (C14:0, C16:0, C18:0) and very-long-chain (C24:0, C24:1) ceramide species and C16:0 glucosylceramide levels were decreased in kidney cortices [39]. In the same study, microarray analysis of kidney cortex from 24-week-old diabetic mice showed significantly altered expression of genes involved in ceramide synthesis and metabolism (decreased expression of Degs2, Cers5, Fads3, Smpd2, and increased expression of Sptlc2, Cers4, S1Pr1, Acer2, Smpdl3b). Similarly, our studies also demonstrated decreased levels of total ceramide in kidney cortices of db/db mice [6,40], where long-chain species (C14:0, C18:0, C18:1, C20:0, C20:1), but not very-long-chain species were affected [6]. In a prospective study on T2D patients from Singapore, longchain ceramide levels (C16:0 and C18:0) were also found elevated in the plasma of patients with early or overt DKD compared to non-DKD controls [41]. Interestingly, high plasma levels of very-long-chain ceramide species were associated with a decreased risk of progression to macroalbuminuria in a subgroup of T1D patients enrolled in the Diabetes Control and Complications Trial (DCCT) and its long-term observational follow-up (EDIC) [42], which may reflect a regulatory role of ceramides in loss of renal function. On the other hand, podocyte-specific deletion of the main catalytic subunit of acid ceramidase resulted in ceramide accumulation in glomeruli and development of nephrotic syndrome in mice [43]. Notably, a recent report on urinary sphingolipids from patients with DKD also showed elevated urinary levels of ceramide d18:1/16:0, d18:1/18:0, d18:0/20:0, d18:1/22:0 and d18:1/24:0, which were correlated with urinary albumin [44]. It should be noted that increased levels of ceramide species were observed in diabetic patients with CKD stage 4, which may be due to the progression of kidney injury and reduced synthesis and/or excretion of ceramides by the kidney. Indeed, production of very-long-chain ceramide species is regulated by ceramide synthases (CerS), where CerS2, the most highly expressed isoform in the kidney, has been found to be associated with albuminuria in diabetic patients [45], and its haploinsufficiency has been reported to induce insulin resistance in the liver of mice fed on a high fat diet (HFD) [46].
In addition, ceramides have gained recent attention due to their role in insulin resistance, a significant contributor to DKD development and progression (as reviewed by us previously in Ref. [47]). Ceramides are known to inhibit signal transduction via phosphatidylinositol-3 kinase and block activation of protein kinase B (AKT), thereby interfering with glucose uptake and impairing storage of glycogen and triglycerides (as reviewed in Ref. [48]). In a recent study using diabetic mice, deletion of dihydroceramide desaturase 1 (DEGS1), an enzyme critical to the formation of ceramide from dihydroceramide, resolved hepatic steatosis and insulin resistance [49]. Similarly, a beneficial effect of pharmacological or genetic ablation of DEGS1 was shown to be associated with decreased obesity-associated lipid accumulation in adipocytes [50]. In a deterministic and stochastic simulator model using cultured macrophages, ceramide and S1P were shown to play a role in controlling the AKT pathway and insulin resistance via manipulating levels of ceramide synthases (CERS2, CERS5 and CERS6) and DEGS2 [51]. In MIN6 cultured cells, elevated levels of ceramides (C14:0, C16:0, C20:1, C24:0) were associated with decreased insulin secretion and increased apoptosis [52], indicating the undeniable involvement of ceramides in development of insulin resistance. The role of ceramides in insulin resistance is reviewed elsewhere in greater detail [53]. Furthermore, we have recently identified a sphingolipid related enzyme that affects insulin receptor (IR) subtype signaling (as reviewed more extensively below).
Ceramide can be further catabolized to sphingosine, which is then phosphorylated to S1P, another important bioactive sphingolipid. Recent studies support an important role for S1P in renal diseases, including renal fibrosis (reviewed in Ref. [54]), nephrotic syndrome [8,9] and other glomerular diseases such as IgA nephropathy and lupus nephritis [55,56]. Early studies of streptozotocin (STZ)-treated rats revealed increased levels of S1P in isolated glomeruli during initial stages of DKD [57]. In support of this observation, another study also demonstrated increased renal levels of S1P in mice with STZ-induced DKD, which was prevented by intraperitoneal injections of insulin [58].
As mentioned earlier, S1P may act as an extra- or intracellular signaling molecule. Extracellular S1P is synthesized in the cytosol by SphK1/Sphk2 and then exported by a number of transporters. Such transporters include SPNS2, MFSD2B, and the ATP-binding cassette (ABC) transporters ABCA, ABCC1, and ABCG2. We previously reported that ABCA1 deficiency is one of the peculiarities in glomeruli isolated from patients with T2D and DKD [2], an observation which we further confirmed by demonstrating that genetic or pharmacological overexpression of ABCA1 is sufficient to ameliorate podocyte injury in a mouse model of DKD [16,17]. Unbalanced expression of S1P receptors (enhanced S1PR2 expression and decreased S1PR1 expression) in mesangial cells of STZ-induced diabetic rats has been also shown to contribute to DKD progression [59]. Increased expression of S1P has been shown to be associated with increased reactive oxygen species production and injury in CKD [60].
On the contrary, much less is known about the role of C1P in DKD development and progression. C1P has been found to modulate AKT phosphorylation in skin fibroblasts, hematopoietic cells [61], macrophages [62,63], and adipocytes [64], consistent with the observation that bioactive sphingolipids are major modulators of insulin signaling [65-67]. Knockdown of ceramide kinase was demonstrated to be efficient in the treatment of mesangioproliferative glomerular diseases [68]. Our studies demonstrated that diabetic db/db mice have less total C1P (with C18:0 species mostly affected) in kidney cortices, and that exogenous administration of C1P protects from DKD progression [6].
It is important to note that other sphingolipids, such as glucosylcerebrosides (GlcCer) and gangliosides (especially GM3), have been previously reported to contribute to DKD development [69-71]. GM3 is the most abundant renal ganglioside. In podocytes, GM3 has been shown to localize in lipid rafts, which are also called GM3-raft domains, and to bind the soluble vascular endothelial growth factor receptor sFLT-1, which plays a critical role in the regulation of angiogenesis and rapid actin reorganization [72]. Moreover, a pivotal role of GM3 in promotion of insulin resistance has been demonstrated [73]. In patients with DKD, increased levels of sialic acid, one of the components of gangliosides, were significantly positively correlated with hemoglobin A1c, serum creatinine, and microalbuminuria [74]. The biology and role of GM3 in diabetes and other metabolic-related diseases is reviewed elsewhere [75]. Levels of glucosylceramide (GlcCer) in diabetic ob/ob mice and STZ-induced diabetic rats are increased in several tissues, including liver, muscle [76] and kidney [69]. Pharmacological inhibition of glycosphingolipid synthesis has been shown to have a beneficial effect on insulin sensitivity and glycemic and weight control in animal models of obesity and diabetes [76-78]. In diabetic db/db mice, elevated levels of hexocyl-, glucosyl-, galactosyl- and lactosylceramides were shown in kidney cortices [79]. In addition, a recent study reported that C18:1 hexosylceramide is associated with DKD, while very-long-chain lactosylceramides are associated with the development of microalbuminuria in patients with T1D [80].
Sphingolipids in FSGS
Compared to DKD, much less is known about the role of sphingolipids in FSGS development and progression. In a mouse model of FSGS (doxorubicin-induced nephropathy), lipid peroxidation was reported as the main driver of macrophage-derived foam cell formation [81]. Using a mass spectrometric metabolomics approach, a recent report demonstrated that the urine of patients with FSGS contains elevated levels of fatty acids (C16:0, C22:4) and lysophosphotidylcholines (C14:0, C18:1) and decreased levels of phosphotidylcholine (C38:4) when compared to healthy subjects [82]. The same study also reported decreased levels of urinary acylcarnitine (C12:0) in patients with FSGS, suggesting impaired fatty acid oxidation and possible mitochondrial dysfunction. Mutations in SGPL1, the gene which encodes S1P lyase, have been reported to be associated with steroid-resistant nephrotic syndrome [8,9,83], which manifests histologically with FSGS and diffuse mesangial sclerosis [84]. Sgpl knockout mice were shown to recapitulate many features of human renal disease [9]. Using microarray analysis on glomeruli isolated from patients with FSGS of the NEPTUNE cohort, we previously demonstrated altered expression of genes involved in cholesterol and free fatty acid homeostasis [4], supporting the hypothesis that dysregulation of cholesterol homeostasis contributes to the pathogenesis of FSGS. Additionally, we previously reported an important role of sphingomyelin phosphodiesterase acid-like 3b (SMPDL3b) in the recurrence of FSGS after transplantation [85]. We demonstrated that the expression of SMPDL3b is significantly decreased in podocytes from patients with recurrent FSGS. The role of SMPDL3b in glomerular diseases will be discussed in detail in the section below.
Therefore, similarly to DKD, modulating the sphingolipid content in glomeruli may also represent a valid strategy to prevent or treat podocyte injury in FSGS.
Role of SMPDL3b in the kidney
SMPDL3b is a glycosylphosphatidylinositol (GPI)- anchored protein [86,87] with reported roles in inflammatory processes [88] and in kidney diseases [6,40,85]. Recently, the crystal structure of murine SMPDL3b was revealed and helped to identify possible substrates for SMPDL3b, which included CDP-choline, ATP and ADP [89]. Sphingomyelin could also be a potential substrate for SMPDL3b.
In macrophages, deficiency of Smpdl3b has been shown to cause alterations in the membrane lipid composition and changes in its fluidity [88,90]. The same study demonstrated that Smpdl3b knockdown results in enhanced responsiveness to Toll-like receptor stimulation (TLR4) and increased inflammatory response. SMPDL3b expression has also been found in pancreatic zymogen granules [91], in plasma protein-depleted cerebrospinal fluid [92], in saliva exosomes [93], in human milk [94], liver [95], and hepatocellular carcinoma [96], suggesting a diverse function of SMPDL3b in different tissues and organs.
In the kidney, we previously reported that SMPDL3b is a resident in lipid raft domains [85]. In the same study, we demonstrated that kidney biopsies of patients with recurrence of FSGS express less SMPDL3b, while overexpression of SMPDL3b in podocytes can prevent actin cytoskeleton disruption and apoptosis. Furthermore, we reported that rituximab, an anti-CD20 monoclonal antibody targeting B cells, binds to SMPDL3b, thereby protecting podocytes from cytoskeleton disruption and apoptosis induced by treatment with sera from patients with recurrent FSGS [85]. Similarly, SMPDL3b protein loss has been reported in xenotransplants, where rituximab pretreatment maintained levels of SMPDL3b [97]. Additionally, adriamycin-induced nephropathy in rats was associated with reduced expression of SMPDL3b, which was also prevented by rituximab [98].
In contrast, SMPDL3b expression is elevated in glomeruli from patients with DKD, as well as in animal models of DKD, where SMPDL3b expression in kidney cortices of 24-week-old diabetic db/db mice was found to be almost three-fold higher when compared to controls using a transcriptomic approach [39]. We previously demonstrated that, in podocytes, SMPDL3b binds to the soluble urokinase plasminogen activator receptor (suPAR) (40), which is reported to be elevated in sera from patients with FSGS and DKD among other causes of CKD [40,99,100]. Furthermore, FSGS sera-treated podocytes showed decreased expression of SMPDL3b in association with increased cortical actin and loss of stress fibers, while podocytes treated with DKD serum demonstrated increased expression of SMPDL3b in association with actin reorganization in cell blebs [40].
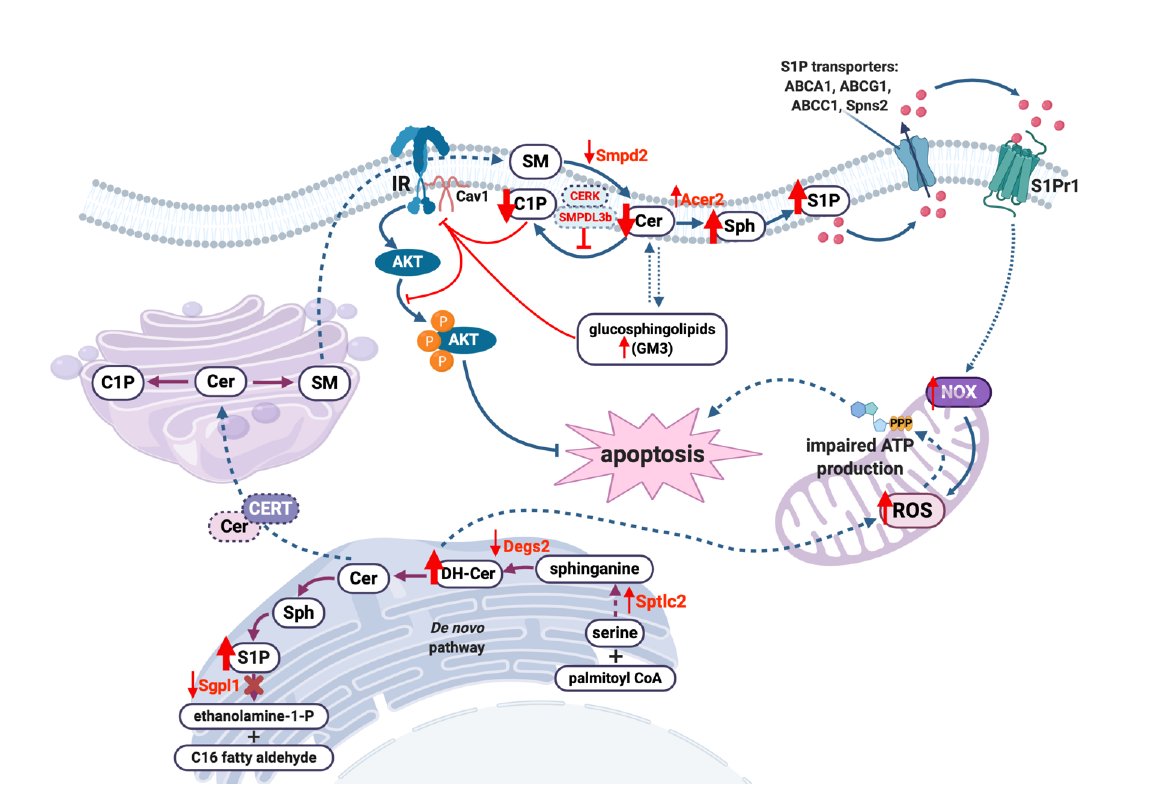
Moreover, an excess of SMPDL3b in human podocytes may cause the displacement of insulin receptor isoforms from caveolin-1-rich domains of the plasma membrane in a C1P-dependent manner, resulting in impaired ability to phosphorylate AKT thus promoting podocyte injury in vitro and development of DKD in vivo [6]. In addition, we demonstrated that overexpression of SMPDL3b in human podocytes causes suppression of protein kinase B and activation of p70S6 kinase phosphorylation, through its binding to both insulin receptor isoforms, IR-A and IR-B and caveolin-1, suggesting a novel role for SMPDL3b as a modulator of insulin signaling in podocytes [6]. Indeed, we demonstrated that SMPDL3b can interfere with the binding of insulin receptor B to caveolin-1 while facilitating insulin receptor A binding to caveolin-1, which may be responsible for increased insulin-stimulated p70S6 kinase phosphorylation. Thereby, overexpression of SMPDL3b is associated with reduced levels of C1P in human podocytes in vitro and in kidney cortices in vivo [6], while podocyte-specific deficiency of SMPDL3b in diabetic db/db mice results in reduced proteinuria and improved renal outcome. The idea that SMPDL3b may regulate the availability of bioactive sphingolipids such as C1P in podocytes is indeed intriguing. However, if SMPDL3b may have a direct phosphatase activity and convert C1P to ceramide remains to be investigated. Our further studies demonstrated that SMPDL3b interacts with ceramide kinase (CERK) and binds C1P in vitro and that SMPDL3b expression positively correlates with CERK expression [7].
Interestingly, single dose irradiation of human podocytes results in a time-dependent decrease of SMPDL3b protein expression in association with cortical actin remodeling, while overexpression of SMPDL3b in human podocytes yields a protective effect from radiation injury [101]. Pretreatment with rituximab mitigated radiation-induced cytoskeletal changes and increased expression levels of SMPDL3b [101]. In contrast, a single dose of radiation applied to human immortalized glomerular endothelial cells results in increased levels of SMPDL3b and decreased levels of C1P [102], while treatment with exogenous C1P or genetic knocking down of SMPDL3b partially protects glomerular endothelial cells. Notably, inhibition of NOX1 seems sufficient to restore normal expression of SMPDL3b and to reduce radiation-induced damage of glomerular endothelial cells. For a more detailed review on the role of sphingolipids in renal oncology, the reader is referred to [103].
Although these reports provide clues to the functions of SMPDL3b in various cells and organs, a better understanding of the role of SMPDL3b in health and disease is still needed. Nevertheless, SMPDL3b seems to be an attractive therapeutic target, at least for the treatment of glomerular diseases.
Targeting Sphingolipids in DKD and FSGS
A role for sphingolipids as modulators of podocyte function in DKD, FSGS and other glomerular diseases is an emerging concept. Increasing evidence suggests an involvement of sphingolipid metabolism in the development and progression of glomerular diseases. It has become clear that targeting sphingolipids may be beneficial for the treatment of DKD and FSGS.
One option to target sphingolipids is through the manipulation of S1P receptors (S1PRs). Treatment with FTY720, or fingolimod, a non-selective S1PR agonist, or SEW2871, a selective agonist of S1PR1, was shown to reduce urinary albumin excretion as well as urinary levels of the pro-inflammatory cytokine tumor necrosis factoralpha in mouse and rat models of STZ-induced DKD [104], suggesting that targeting kidney S1PR1 may represent a therapeutic intervention for the treatment of DKD. Additionally, treatment with berberine improved renal injury in STZ-induced diabetic rats via downregulation of S1PR2 [105] and the use of the S1PR2 specific antagonist LTE-013 showed improved insulin resistance in human and rat hepatocytes [106], suggesting the possibility that S1PR2 inhibition may prove useful for the treatment of diabetes and its related complications. Another study demonstrated that inhibition of ceramide accumulation with myriocin in Otsuka Long Evans Tokushima fatty rats and HFD-fed mice ameliorates albuminuria and histologic features of DKD [107]. Finally, our studies demonstrated that exogenous C1P treatment or podocyte-specific inhibition of SMDP3Lb results in reduced proteinuria, improves renal outcome and restores insulin receptor signaling in diabetic db/db mice [6].
While the therapeutic use of active sphingolipids or modulators of S1P receptor activation remains of high interest and deserves further study, the possibility to target sphingolipid related enzymes remains an attractive opportunity. Among them, SMPDL3b may surely represent an attractive target. As no drug is yet available to directly agonize and antagonize the function of SMPDL3b, repurposing strategies with rituximab or other anti-CD20 antibodies that recognize the same epitope could and should be considered. Our retrospective study clearly suggested that a single dose of rituximab administered in the pre-transplant setting may be sufficient to prevent the recurrence of proteinuria after transplantation in patients with FSGS [85]. Observational studies have demonstrated that rituximab is a safe and effective treatment option in patients with steroid-dependent or frequently relapsing nephrotic syndrome, including minimal change disease and FSGS. While randomized controlled trials are needed, rituximab has been shown to prevent recurrences and reduce the need for immunosuppressive therapy [108,109]. A recent meta-analysis on the use of rituximab in clinical settings suggests an additional benefit of rituximab if added to the standard therapy in adults with FSGS [110]. Therefore, rituximab may represent a potentially effective agent for the treatment of some glomerular diseases. Whether these beneficial effects are due to B lymphocyte depletion or to stabilization of SMPDL3b expression and function cannot be discerned in treated patients and requires further experimental studies with humanized mouse models.
Another reported therapy targeting sphingolipids is enzyme replacement therapy (ERT), which currently represents a standard of therapy for Fabry disease, or lipid storage disease. In a recent case report of a Japanese man with FSGS and low activity of alpha-galactosidase (with mutation in M296I), ERT in association with immunotherapy with steroids and cyclosporine A [111] improved proteinuria levels, while in another case report of an obese male with histologically proven FSGS and low activity of alpha-galactosidase (with missense mutation in R310Q) no improvement in renal function despite ERT was described [112]. Thus, while ERT may be a very promising therapy for the treatment of lipid storage disease, its therapeutic potential in the treatment of patients with FSGS warrants further investigation.
Concluding Remarks
In this review, we highlighted new research trends and scientific knowledge acquired within the past few years indicating that sphingolipids are key players contributing to the pathogenesis and progression of glomerular diseases such as DKD and FSGS (Figure 2). Studies suggest that SMPDL3b, S1P lyase, C1P, S1P and S1P receptors are valid and important targets for the development of novel therapeutic therapies for glomerular diseases. Manipulation of the S1P signaling pathway, particularly modulation of S1P receptors and/or sphingosine kinases is evolving as an attractive therapeutic strategy to slow the progression of kidney diseases. An important question, which needs to be further addressed in detail, is the contribution of altered sphingolipid metabolism to the pathogenesis of glomerular diseases. Indeed, while many reports describe changes in sphingolipid levels and species at disease onset, it remains unclear if these changes are in fact pathogenic. Moreover, because of their biophysical properties, sphingolipids are less capable of moving freely from one compartment to another inside of a cell. Thus, a better understanding of the relationship between the localization and function of sphingolipids in different cell compartments is needed as it may explain some of the conflicting reports in the literature and further our understanding of the role of sphingolipids in the pathogenesis and progression of glomerular diseases.
Conflicts of Interest
A.F. and S.M. are inventors on pending or issued patents (PCT/US11/56272, PCT/US12/62594, PCT/ US2019/041730, PCT/US2019/032215, PCT/US13/36484 and PCT 62/674,897) aimed to diagnosing or treating proteinuric kidney diseases. They stand to gain royalties from their future commercialization of these patents. A.F. is Vice-President of L&F Health LLC and is consultant for ZyVersa Therapeutics, Inc. ZyVersa Therapeutics, Inc has licensed worldwide rights to develop and commercialize hydroxypropyl-beta-cyclodextrin from L&F Research for the treatment of kidney disease. A.F. is founder of LipoNexT LLC. S.M. is a consultant for Kintai Therapeutics, Inc and holds equity interest in L&F Research. AF and SM are supported by Boehringer Ingelheim. AM and YD declare no conflicts of interest.
Funding Statement
AF and SM are supported by NIH grants R01DK117599, R01DK104753, and R01CA227493, and by Boehringer Ingelheim. AF is supported by U54DK083912, UM1DK100846, U01DK116101, and UL1TR000460 (Miami Clinical and Translational Science Institute).
Acknowledgments
We give a special thanks to the Katz family for continuous support. We would like to acknowledge the BioRender team for creating user-friendly and simple software to use for creating of illustration (www.biorender.com).
Author Contribution
All authors listed have made a substantial, direct and intellectual contribution to the work. AM prepared the draft of manuscript and the figures; YD, SM and AF reviewed and edited the manuscript.
References
2. Merscher-Gomez S, Guzman J, Pedigo CE, Lehto M, Aguillon-Prada R, Mendez A, et al. Cyclodextrin protects podocytes in diabetic kidney disease. Diabetes. 2013 Nov 1;62(11):3817-27.
3. Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. Journal of Lipid Research. 2014 Mar 1;55(3):561-72.
4. Mitrofanova A, Molina J, Varona Santos J, Guzman J, Morales XA, Ducasa GM, et al. Hydroxypropyl-β-cyclodextrin protects from kidney disease in experimental Alport syndrome and focal segmental glomerulosclerosis. Kidney International. 2018 Dec 1;94(6):1151-9.
5. Bjercke O. The Norwegian Medical Association as it is. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. 1970 May 30;90(106):Suppl-1180.
6. Mitrofanova A, Mallela SK, Ducasa GM, Yoo TH, Rosenfeld-Gur E, Zelnik ID, et al. SMPDL3b modulates insulin receptor signaling in diabetic kidney disease. Nature Communications. 2019 Jun 19;10(1):1-6.
7. Mallela SK, Mitrofanova A, Merscher S, Fornoni A. Regulation of the amount of ceramide-1-phosphate synthesized in differentiated human podocytes. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2019 Dec 1;1864(12):158517.
8. Lovric S, Goncalves S, Gee HY, Oskouian B, Srinivas H, Choi WI, et al. Mutations in sphingosine-1-phosphate lyase cause nephrosis with ichthyosis and adrenal insufficiency. The Journal of Clinical Investigation. 2017 Mar 1;127(3):912-28.
9. Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, et al. Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. The Journal of Clinical Investigation. 2017 Mar 1;127(3):942-53.
10. Schumann J, Grevot A, Ledieu D, Wolf A, Schubart A, Piaia A, et al. Reduced activity of sphingosine-1-phosphate lyase induces podocyte-related glomerular proteinuria, skin irritation, and platelet activation. Toxicologic Pathology. 2015 Jul;43(5):694-703.
11. Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, et al. Deficiency of the sphingosine- 1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Human Mutation. 2017 Apr;38(4):365-72.
12. Chronic Kidney Disease in the United States [Internet]. US Department of Health and Human Services, Centers for Disease Control and Prevention. 2019 [cited May 13, 2020]. Available from: https://www.cdc.gov/kidneydisease/pdf/2019_National-Chronic-Kidney-Disease- Fact-Sheet.pdf.
13. Pagtalunan ME, Miller PL, Jumping-Eagle S, Nelson RG, Myers BD, Rennke HG, et al. Podocyte loss and progressive glomerular injury in type II diabetes. The Journal of Clinical Investigation. 1997 Jan 15;99(2):342-8.
14. Toyoda M, Najafian B, Kim Y, Caramori ML, Mauer M. Podocyte detachment and reduced glomerular capillary endothelial fenestration in human type 1 diabetic nephropathy. Diabetes. 2007;56(8):2155-60.
15. Tang C, Kanter JE, Bornfeldt KE, Leboeuf RC, Oram JF. Diabetes reduces the cholesterol exporter ABCA1 in mouse macrophages and kidneys. Journal of Lipid Research. 2010 Jul 1;51(7):1719-28.
16. Pedigo CE, Ducasa GM, Leclercq F, Sloan A, Mitrofanova A, Hashmi T, et al. Local TNF causes NFATc1- dependent cholesterol-mediated podocyte injury. The Journal of Clinical Investigation. 2016 Sep 1;126(9):3336- 50.
17. Ducasa GM, Mitrofanova A, Mallela SK, Liu X, Molina J, Sloan A, et al. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. The Journal of Clinical Investigation. 2019 Jul 22;129(8).
18. O’Shaughnessy MM, Hogan SL, Poulton CJ, Falk RJ, Singh HK, Nickeleit V, et al. Temporal and demographic trends in glomerular disease epidemiology in the Southeastern United States, 1986–2015. Clinical Journal of the American Society of Nephrology. 2017 Apr 3;12(4):614-23.
19. Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, et al. Estimating podocyte number and density using a single histologic section. Journal of the American Society of Nephrology. 2014 May 1;25(5):1118- 29.
20. Futerman AH, Hannun YA. The complex life of simple sphingolipids. EMBO reports. 2004 Aug;5(8):777-82.
21. Bieberich E. Sphingolipids and lipid rafts: Novel concepts and methods of analysis. Chemistry and Physics of Lipids. 2018 Nov 1;216:114-31.
22. Moro K, Kawaguchi T, Tsuchida J, Gabriel E, Qi Q, Yan L, et al. Ceramide species are elevated in human breast cancer and are associated with less aggressiveness. Oncotarget. 2018 Apr 13;9(28):19874.
23. Gault CR, Obeid LM, Hannun YA. An overview of sphingolipid metabolism: from synthesis to breakdown. Advances in Experimental Medicine and Biology. 2010;688:1-23.
24. Gomez-Munoz A, Duffy PA, Martin A, O’Brien L, Byun HS, Bittman R, et al. Short-chain ceramide-1-phosphates are novel stimulators of DNA synthesis and cell division: antagonism by cell-permeable ceramides. Molecular Pharmacology. 1995 May 1;47(5):833-9.
25. Gomez-Munoz A, Kong JY, Salh B, Steinbrecher UP. Ceramide-1-phosphate blocks apoptosis through inhibition of acid sphingomyelinase in macrophages. Journal of Lipid Research. 2004 Jan 1;45(1):99-105.
26. Presa N, Gomez-Larrauri A, Dominguez-Herrera A, Trueba M, Gomez-Munoz A. Novel signaling aspects of ceramide 1-phosphate. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids. 2020 Apr 1;1865(4):158630.
27. Simanshu DK, Kamlekar RK, Wijesinghe DS, Zou X, Zhai X, Mishra SK, et al. Non-vesicular trafficking by a ceramide-1-phosphate transfer protein regulates eicosanoids. Nature. 2013 Aug;500(7463):463-7.
28. Gómez-Muñoz A. Ceramide 1-phosphate/ceramide, a switch between life and death. Biochimica et Biophysica Acta (BBA)-Biomembranes. 2006 Dec 1;1758(12):2049- 56.
29. Blom T, Bergelin N, Meinander A, Löf C, Slotte JP, Eriksson JE, et al. An autocrine sphingosine-1-phosphate signaling loop enhances NF-κB-activation and survival. BMC cell biology. 2010 Dec;11(1):1-1.
30. Yaghobian D, Don AS, Yaghobian S, Chen X, Pollock CA, Saad S. Increased sphingosine 1-phosphate mediates inflammation and fibrosis in tubular injury in diabetic nephropathy. Clinical and Experimental Pharmacology and Physiology. 2016 Jan;43(1):56-66.
31. Ramanathan R, Raza A, Sturgill J, Lyon D, Young J, Hait NC, et al. Paradoxical association of postoperative plasma sphingosine-1-phosphate with breast cancer aggressiveness and chemotherapy. Mediators of Inflammation. 2017 Sep 24;2017:5984819.
32. Nakajima M, Nagahashi M, Rashid OM, Takabe K, Wakai T. The role of sphingosine-1-phosphate in the tumor microenvironment and its clinical implications. Tumor Biology. 2017 Apr;39(4):1010428317699133.
33. Winkler MS, Nierhaus A, Mudersbach E, Holzmann M, Bauer A, Robbe L, et al. Sphingosine-1-phosphate is a new biomarker for severity in human sepsis. Critical Care. 2015 Dec;19(1):1-201.
34. Hsu SC, Chang JH, Hsu YP, Bai KJ, Huang SK, Hsu CW. Circulating sphingosine-1-phosphate as a prognostic biomarker for community-acquired pneumonia. PloS One. 2019 May 15;14(5):e0216963.
35. Merscher S, Fornoni A. Podocyte pathology and nephropathy–sphingolipids in glomerular diseases. Frontiers in Endocrinology. 2014 Jul 30;5:127.
36. Kremer GJ, Atzpodien W, Schnellbacher E. Plasma glycosphingolipids in diabetics and normals. Klinische Wochenschrift. 1975 Jul 1;53(13):637-8.
37. Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, et al. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes. 2009 Feb 1;58(2):337-43.
38. Gorska M, Dobrzyn A, Baranowski M. Concentrations of sphingosine and sphinganine in plasma of patients with type 2 diabetes. Medical Science Monitor. 2005 Jan 1;11(1):CR35-8.
39. Sas KM, Nair V, Byun J, Kayampilly P, Zhang H, Saha J, et al. Targeted lipidomic and transcriptomic analysis identifies dysregulated renal ceramide metabolism in a mouse model of diabetic kidney disease. Journal of Proteomics & Bioinformatics. 2015 Oct: Suppl 14.
40. Yoo TH, Pedigo CE, Guzman J, Correa-Medina M, Wei C, Villarreal R, et al. Sphingomyelinase-like phosphodiesterase 3b expression levels determine podocyte injury phenotypes in glomerular disease. Journal of the American Society of Nephrology. 2015 Jan 1;26(1):133-47.
41. Liu JJ, Ghosh S, Kovalik JP, Ching J, Choi HW, Tavintharan S, et al. Profiling of plasma metabolites suggests altered mitochondrial fuel usage and remodeling of sphingolipid metabolism in individuals with type 2 diabetes and kidney disease. Kidney International Reports. 2017 May 1;2(3):470-80.
42. Klein RL, Hammad SM, Baker NL, Hunt KJ, Al Gadban MM, Cleary PA, et al. Decreased plasma levels of select very long chain ceramide species are associated with the development of nephropathy in type 1 diabetes. Metabolism. 2014 Oct 1;63(10):1287-95.
43. Li G, Kidd J, Kaspar C, Dempsey S, Bhat OM, Camus S, et al. Podocytopathy and Nephrotic Syndrome in Mice with Podocyte-Specific Deletion of the Asah1 Gene: Role of Ceramide Accumulation in Glomeruli. The American Journal of Pathology. 2020 Mar 16.
44. Morita Y, Kurano M, Sakai E, Nishikawa T, Nishikawa M, Sawabe M, et al. Analysis of urinary sphingolipids using liquid chromatography-tandem mass spectrometry in diabetic nephropathy. Journal of Diabetes Investigation. 2020 Mar;11(2):441-9.
45. Shiffman D, Pare G, Oberbauer R, Louie JZ, Rowland CM, Devlin JJ, et al. A gene variant in CERS2 is associated with rate of increase in albuminuria in patients with diabetes from ONTARGET and TRANSCEND. PLoS One. 2014 Sep 19;9(9):e106631.
46. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, et al. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metabolism. 2014 Oct 7;20(4):687- 95.
47. Mitrofanova A, Sosa MA, Fornoni A. Lipid mediators of insulin signaling in diabetic kidney disease. American Journal of Physiology-Renal Physiology. 2019 Nov 1;317(5):F1241-52.
48. Chaurasia B, Summers SA. Ceramides–lipotoxic inducers of metabolic disorders. Trends in Endocrinology & Metabolism. 2015 Oct 1;26(10):538-50.
49. Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, Wang L, et al. Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science. 2019 Jul 26;365(6451):386-92.
50. Barbarroja N, Rodriguez-Cuenca S, Nygren H, Camargo A, Pirraco A, Relat J, et al. Increased dihydroceramide/ceramide ratio mediated by defective expression of degs1 impairs adipocyte differentiation and function. Diabetes. 2015 Apr 1;64(4):1180-92.
51. Reali F, Morine MJ, Kahramanoğulları O, Raichur S, Schneider H-C, Crowther D, et al. Mechanistic interplay between ceramide and insulin resistance. Scientific Reports. 2017 Jan 23;7(1):1-9.
52. Manukyan L, Ubhayasekera SJ, Bergquist J, Sargsyan E, Bergsten P. Palmitate-induced impairments of β-cell function are linked with generation of specific ceramide species via acylation of sphingosine. Endocrinology. 2015 Mar 1;156(3):802-12.
53. Sokolowska E, Blachnio-Zabielska A. The Role of Ceramides in Insulin Resistance. Front Endocrinol (Lausanne). 2019;10:577.
54. Zhang X, Ritter JK, Li N. Sphingosine-1-phosphate pathway in renal fibrosis. American Journal of Physiology- Renal Physiology. 2018 Oct 1;315(4):F752-6.
55. Su K, Zeng P, Liang W, Luo Z, Wang Y, Lv X, et al. FTY720 Attenuates Angiotensin II- Induced Podocyte Damage via Inhibiting Inflammatory Cytokines. Mediators of inflammation. 2017 Feb 7;2017:3701385.
56. Okazaki H, Hirata D, Kamimura T, Sato H, Iwamoto M, Yoshio T, et al. Effects of FTY720 in MRL-lpr/lpr mice: therapeutic potential in systemic lupus erythematosus. The Journal of Rheumatology. 2002 Apr 1;29(4):707-16.
57. Geoffroy K, Troncy L, Wiernsperger N, Lagarde M, El Bawab S. Glomerular proliferation during early stages of diabetic nephropathy is associated with local increase of sphingosine-1- phosphate levels. FEBS Letters. 2005 Feb 14;579(5):1249-54.
58. Nojiri T, Kurano M, Tokuhara Y, Ohkubo S, Hara M, Ikeda H, et al. Modulation of sphingosine-1-phosphate and apolipoprotein M levels in the plasma, liver and kidneys in streptozotocin-induced diabetic mice. Journal of Diabetes Investigation. 2014 Nov;5(6):639-48.
59. Imasawa T, Kitamura H, Ohkawa R, Satoh Y, Miyashita A, Yatomi Y. Unbalanced expression of sphingosine 1-phosphate receptors in diabetic nephropathy. Experimental and Toxicologic Pathology. 2010 Jan 1;62(1):53-60.
60. Bhat OM, Yuan X, Li G, Lee R, Li PL. Sphingolipids and Redox Signaling in Renal Regulation and Chronic Kidney Diseases. Antioxidants & Redox Signaling. 2018 Apr 1;28(10):1008-26.
61. Kim CH, Wu W, Wysoczynski M, Abdel-Latif A, Sunkara M, Morris A, et al. Conditioning for hematopoietic transplantation activates the complement cascade and induces a proteolytic environment in bone marrow: a novel role for bioactive lipids and soluble C5b-C9 as homing factors. Leukemia. 2012;26(1):106-16.
62. Gomez-Munoz A, Kong JY, Parhar K, Wang SW, Gangoiti P, Gonzalez M, et al. Ceramide- 1-phosphate promotes cell survival through activation of the phosphatidylinositol 3-kinase/protein kinase B pathway. FEBS letters. 2005 Jul 4;579(17):3744-50.
63. Gangoiti P, Granado MH, Wang SW, Kong JY, Steinbrecher UP, Gomez-Munoz A. Ceramide 1-phosphate stimulates macrophage proliferation through activation of the PI3- kinase/PKB, JNK and ERK1/2 pathways. Cellular Signalling. 2008 Apr 1;20(4):726-36.
64. Chaurasia B, Kaddai VA, Lancaster GI, Henstridge DC, Sriram S, Galam DL, et al. Adipocyte Ceramides Regulate Subcutaneous Adipose Browning, Inflammation, and Metabolism. Cell Metabolism. 2016 Dec 13;24(6):820-34.
65. Summers SA. Sphingolipids and insulin resistance: the five Ws. Current Opinion in Lipidology. 2010 Apr 1;21(2):128-35.
66. Meikle PJ, Summers SA. Sphingolipids and phospholipids in insulin resistance and related metabolic disorders. Nature Reviews Endocrinology. 2017 Feb;13(2):79.
67. Jesko H, Stepien A, Lukiw WJ, Strosznajder RP. The Cross-Talk Between Sphingolipids and Insulin-Like Growth Factor Signaling: Significance for Aging and Neurodegeneration. Molecular Neurobiology. 2019 May 1;56(5):3501-21.
68. Pastukhov O, Schwalm S, Romer I, Zangemeister- Wittke U, Pfeilschifter J, Huwiler A. Ceramide kinase contributes to proliferation but not to prostaglandin E2 formation in renal mesangial cells and fibroblasts. Cellular Physiology and Biochemistry: International Journal of Experimental Cellular Physiology, Biochemistry, and Pharmacology. 2014;34(1):119-33.
69. Zador IZ, Deshmukh GD, Kunkel R, Johnson K, Radin NS, Shayman JA. A role for glycosphingolipid accumulation in the renal hypertrophy of streptozotocininduced diabetes mellitus. The Journal of clinical investigation. 1993 Mar 1;91(3):797-803.
70. Kwak DH, Rho YI, Kwon OD, Ahan SH, Song JH, Choo YK, et al. Decreases of ganglioside GM3 in streptozotocininduced diabetic glomeruli of rats. Life Sciences. 2003 Mar 14;72(17):1997-2006.
71. Novak A, Režić Mužinić N, Cikeš Čulić V, Božić J, Tičinović Kurir T, Ferhatović L, et al. Renal distribution of ganglioside GM3 in rat models of types 1 and 2 diabetes. Journal of Physiology and Biochemistry. 2013 Dec 1;69(4):727-35.
72. Jin J, Sison K, Li C, Tian R, Wnuk M, Sung HK, et al. Soluble FLT1 binds lipid microdomains in podocytes to control cell morphology and glomerular barrier function. Cell. 2012 Oct 12;151(2):384-99.
73. Kabayama K, Sato T, Saito K, Loberto N, Prinetti A, Sonnino S, et al. Dissociation of the insulin receptor and caveolin-1 complex by ganglioside GM3 in the state of insulin resistance. Proceedings of the National Academy of Sciences. 2007 Aug 21;104(34):13678-83.
74. Ene CD, Penescu M, Anghel A, Neagu M, Budu V, Nicolae I. Monitoring Diabetic Nephropathy by Circulating Gangliosides. Journal of Immunoassay and Immunochemistry. 2016 Jan 2;37(1):68-79.
75. Inokuchi JI, Inamori KI, Kabayama K, Nagafuku M, Uemura S, Go S, et al. Progress in Molecular Biology and Translational Science 2018 Jan 1;156:151-95.
76. Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, et al. Pharmacological Inhibition of Glucosylceramide Synthase Enhances Insulin Sensitivity. Diabetes. 2007 May 1;56(5):1341-9.
77. Zhao H, Przybylska M, Wu I-H, Zhang J, Siegel C, Komarnitsky S, et al. Inhibiting Glycosphingolipid Synthesis Improves Glycemic Control and Insulin Sensitivity in Animal Models of Type 2 Diabetes. Diabetes. 2007 May 1;56(5):1210-8.
78. Jang HJ, Lim S, Kim JM, Yoon S, Lee CY, Hwang HJ, et al. Glucosylceramide synthase regulates adipo-osteogenic differentiation through synergistic activation of PPARγ with GlcCer. The FASEB Journal. 2020 Jan;34(1):1270- 87.
79. Subathra M, Korrapati M, Howell LA, Arthur JM, Shayman JA, Schnellmann RG, et al. Kidney glycosphingolipids are elevated early in diabetic nephropathy and mediate hypertrophy of mesangial cells. American Journal of Physiology-Renal Physiology. 2015 Aug 1;309(3):F204-15.
80. Lopes-Virella MF, Baker NL, Hunt KJ, Hammad SM, Arthur J, Virella G, et al. Glycosylated sphingolipids and progression to kidney dysfunction in type 1 diabetes. Journal of Clinical Lipidology. 2019 May 1;13(3):481-91.
81. Hara S, Kobayashi N, Sakamoto K, Ueno T, Manabe S, Takashima Y, et al. Podocyte injury-driven lipid peroxidation accelerates the infiltration of glomerular foam cells in focal segmental glomerulosclerosis. The American Journal of Pathology. 2015 Aug 1;185(8):2118-31.
82. Erkan E, Zhao X, Setchell K, Devarajan P. Distinct urinary lipid profile in children with focal segmental glomerulosclerosis. Pediatric Nephrology. 2016 Apr 1;31(4):581-8.
83. Linhares ND, Arantes RR, Araujo SA, Pena SDJ. Nephrotic syndrome and adrenal insufficiency caused by a variant in SGPL1. Clinical kidney journal. 2018 Aug;11(4):462-7.
84. Kemper MJ, Lemke A. Treatment of Genetic Forms of Nephrotic Syndrome. Frontiers in Pediatrics. 2018 Mar 26;6:72.
85. Fornoni A, Sageshima J, Wei C, Merscher-Gomez S, Aguillon-Prada R, Jauregui AN, et al. Rituximab targets podocytes in recurrent focal segmental glomerulosclerosis. Science Translational Medicine. 2011 Jun 1;3(85):85ra46.
86. Masuishi Y, Nomura A, Okayama A, Kimura Y, Arakawa N, Hirano H. Mass spectrometric identification of glycosylphosphatidylinositol-anchored peptides. Journal of Proteome rResearch. 2013 Oct 4;12(10):4617-26.
87. Cortes LK, Vainauskas S, Dai N, McClung CM, Shah M, Benner JS, et al. Proteomic identification of mammalian cell surface derived glycosylphosphatidylinositol-anchored proteins through selective glycan enrichment. Proteomics. 2014 Nov;14(21-22):2471-84.
88. Heinz LX, Baumann CL, Köberlin MS, Snijder B, Gawish R, Shui G, et al. The Lipid- Modifying Enzyme SMPDL3B Negatively Regulates Innate Immunity. Cell Reports. 2015 Jun 30;11(12):1919-28.
89. Gorelik A, Heinz LX, Illes K, Superti-Furga G, Nagar B. Crystal Structure of the Acid Sphingomyelinase-like Phosphodiesterase SMPDL3B Provides Insights into Determinants of Substrate Specificity. Journal of Biological Chemistry. 2016 Nov 11;291(46):24054-64.
90. Köberlin MS, Snijder B, Heinz LX, Baumann CL, Fauster A, Vladimer GI, et al. A Conserved Circular Network of Coregulated Lipids Modulates Innate Immune Responses. Cell. 2015 Jul 2;162(1):170-83.
91. Rindler MJ, Xu CF, Gumper I, Smith NN, Neubert TA. Proteomic analysis of pancreatic zymogen granules: identification of new granule proteins. Journal of Proteome Research. 2007 Aug 3;6(8):2978-92.
92. Thouvenot E, Urbach S, Dantec C, Poncet J, Séveno M, Demettre E, et al. Enhanced detection of CNS cell secretome in plasma protein-depleted cerebrospinal fluid. Journal of Proteome Research. 2008 Oct 3;7(10):4409-21.
93. Ogawa Y, Miura Y, Harazono A, Kanai-Azuma M, Akimoto Y, Kawakami H, et al. Proteomic analysis of two types of exosomes in human whole saliva. Biological and Pharmaceutical Bulletin. 2011 Jan 1;34(1):13-23.
94. Picariello G, Ferranti P, Mamone G, Klouckova I, Mechref Y, Novotny MV, et al. Gel-free shotgun proteomic analysis of human milk. Journal of Chromatography A. 2012 Mar 2;1227:219-33.
95. Reichel M, Rhein C, Hofmann LM, Monti J, Japtok L, Langgartner D, et al. Chronic Psychosocial Stress in Mice Is Associated With Increased Acid Sphingomyelinase Activity in Liver and Serum and With Hepatic C16:0- Ceramide Accumulation. Frontiers in Psychiatry. 2018 Oct 16;9:496.
96. Liu B, Xiao J, Dong M, Qiu Z, Jin J. Human alkaline ceramidase 2 promotes the growth, invasion, and migration of hepatocellular carcinoma cells via sphingomyelin phosphodiesterase acid-like 3B. Cancer Science. 2020 May 23.
97. Tasaki M, Shimizu A, Hanekamp I, Torabi R, Villani V, Yamada K. Rituximab treatment prevents the early development of proteinuria following pig-to-baboon xenokidney transplantation. Journal of the American Society of Nephrology. 2014 Apr 1;25(4):737-44.
98. Takahashi Y, Ikezumi Y, Saitoh A. Rituximab protects podocytes and exerts anti- proteinuric effects in rat adriamycin-induced nephropathy independent of B-lymphocytes. Nephrology. 2017 Jan;22(1):49-57.
99. Hayek SS, Sever S, Ko YA, Trachtman H, Awad M, Wadhwani S, et al. Soluble Urokinase Receptor and Chronic Kidney Disease. New England Journal of Medicine. 2015 Nov 12;373(20):1916-25.
100. Wei C, El Hindi S, Li J, Fornoni A, Goes N, Sageshima J, et al. Circulating urokinase receptor as a cause of focal segmental glomerulosclerosis. Nature Medicine. 2011 Aug;17(8):952-60.
101. Ahmad A, Mitrofanova A, Bielawski J, Yang Y, Marples B, Fornoni A, et al. Sphingomyelinase-like phosphodiesterase 3b mediates radiation-induced damage of renal podocytes. The FASEB Journal. 2017 Feb;31(2):771-80.
102. Abou Daher A, Francis M, Azzam P, Ahmad A, Eid AA, Fornoni A, et al. Modulation of radiation-induced damage of human glomerular endothelial cells by SMPDL3B. The FASEB Journal. 2020 Apr 15.
103. Abou Daher A, El Jalkh T, Eid AA, Fornoni A, Marples B, Zeidan YH. Translational Aspects of Sphingolipid Metabolism in Renal Disorders. International Journal of Molecular Sciences. 2017 Dec;18(12):2528.
104. Awad AS, Rouse MD, Khutsishvili K, Huang L, Bolton WK, Lynch KR, et al. Chronic sphingosine 1-phosphate 1 receptor activation attenuates early-stage diabetic nephropathy independent of lymphocytes. Kidney International. 2011 May 2;79(10):1090-8.
105. Huang K, Liu W, Lan T, Xie X, Peng J, Huang J, et al. Berberine reduces fibronectin expression by suppressing the S1P-S1P2 receptor pathway in experimental diabetic nephropathy models. PLoS One. 2012 Aug 24;7(8):e43874.
106. Fayyaz S, Henkel J, Japtok L, Krämer S, Damm G, Seehofer D, et al. Involvement of sphingosine 1-phosphate in palmitate-induced insulin resistance of hepatocytes via the S1P2 receptor subtype. Diabetologia. 2014 Feb 1;57(2):373-82.
107. Woo CY, Baek JY, Kim AR, Hong CH, Yoon JE, Kim HS, et al. Inhibition of Ceramide Accumulation in Podocytes by Myriocin Prevents Diabetic Nephropathy. Diabetes & Metabolism Journal. 2019 Jan 3;43.
108. Ruggenenti P, Ruggiero B, Cravedi P, Vivarelli M, Massella L, Marasà M, et al. Rituximab in Steroid- Dependent or Frequently Relapsing Idiopathic Nephrotic Syndrome. Journal of the American Society of Nephrology. 2014 Apr 1;25(4):850-63.
109. Munyentwali H, Bouachi K, Audard V, Remy P, Lang P, Mojaat R, et al. Rituximab is an efficient and safe treatment in adults with steroid-dependent minimal change disease. Kidney International. 2013 Mar 1;83(3):511-6.
110. Hansrivijit P, Cheungpasitporn W, Thongprayoon C, Ghahramani N. Rituximab therapy for focal segmental glomerulosclerosis and minimal change disease in adults: a systematic review and meta-analysis. BMC Nephrology. 2020 Dec;21(1):134.
111. Fujisawa H, Nakayama Y, Nakao S, Yamamoto R, Kurokawa Y, Nakamura N, et al. Effectiveness of immunosuppressive therapy for nephrotic syndrome in a patient with late-onset Fabry disease: a case report and literature review. BMC Nephrology. 2019 Dec 1;20(1):469.
112. Saito A, Kimura T, Takeuchi Y, Matsuda K, Fukami H, Sato H, et al. A case of rapid progression of Fabry nephropathy with remarkable glomerulomegaly: a case report and mini literature review of weak response to enzyme replacement therapy (ERT). Renal Replacement Therapy. 2016 Dec 1;2(1):69.