Main Text
Inflammatory processes are essential for tissue integrity in living systems as they ward off intruders such as parasites, microbes and viruses [1], rendering them protective safeguards to control external threats. However, inflammation many times is accompanied by collateral tissue damage, and is thus followed by tightly orchestrated phases of recovery [1]. Indeed, these mechanisms are not only operational in peripheral tissue, but also occur within the central nervous system (CNS). Indeed, proper function of brain and spinal cord is of utmost importance to an organism, as they control and regulate all vital organs to concert proper function of the entire system. Strict regulation of inflammation is therefore particularly important here, as damage is especially destructive [2]. Disruption of the delicate balance of targeted inflammatory destruction and tissue recovery can lead to chronic or unregulated immune inflammatory cascades. Importantly, this balance is challenged especially in neuroinflammatory and degenerative diseases of the CNS, such as Alzheimer’s disease (AD), Parkinson’s disease (PD) and Multiple Sclerosis (MS) [2]. These pathological conditions are characterized by chronic inflammation and dysregulated recovery mechanisms, leading to long-term tissue damage and functional loss [3]. Key players of inflammatory tissue damage include CNS-infiltrating T cells, neutrophils and macrophages. However, recent research has revealed that CNS resident cells including astrocytes, microglia, and oligodendrocytes are involved crucially in the regulation of these cascades [2].
Microglia are the CNS resident macrophage population, accounting for up to 10% of the total cell population of the CNS [4]. Despite their comparatively small proportion, they are characterized by pronounced morphological and functional heterogeneity, which is strongly influenced by their local microenvironment [5]. For a long time, activated microglia were primarily considered to be drivers of neuronal damage particularly during chronic inflammatory neurodegenerative processes [4,6]. However, it has become evident in recent years that microglia play a central role in maintaining neuronal function in the healthy CNS [7]: microglia are in close contact with neurons, monitor the local microenvironment with their processes actively and react to even minor changes in the extracellular environment by several mechanisms [8]. Studies in animal models show that a lack of functional microglia leads to increased neuronal damage after toxic or ischemic insults, underscoring their necessity for neuroprotective functions [9,10]. In addition to their monitoring functions, microglia share numerous properties with other mononuclear phagocytes, including cytokine secretion and limited antigen presentation [4,11]. Moreover, microglia are heavily involved in shaping neuronal synaptic plasticity, neurotransmitter recycling, and removal of synapses and debris, thus influencing complex processes such as behavior and learning [12,13].
Depending on tissue-intrinsic and -extrinsic cues, microglia exert both pro- and anti-inflammatory, as well as tissue-destructive or regenerative effects: they produce a variety of soluble factors, are involved in the removal of dead cells and contribute to the protection of neurons [7,14]. In parallel, microglia actively restrain excessive and tissue-damaging immune responses, for instance by promoting the expression of inflammation-resolving factors such as Interleukin (IL)-10 or Transforming Growth Factor β (TGFβ) or by inducing apoptosis of infiltrating T cells [4,15,16]. This functional versatility highlights the importance of microglia for CNS homeostasis and health. However, pathological conditions such as MS, PD or AD, are characterized by microglial activation and microglia-mediated or -enhanced tissue damage (Figure 1). Indeed, their inflammatory activation leads to an increase in proliferation, but also enhanced ability of antigen presentation and secretion of pro-inflammatory mediators, such as Tumor necrosis factor α (TNFα), IL1β or reactive oxygen species, ultimately shaping pro-inflammatory and tissue-destructive microenvironment [17,18]. Even though this inflammatory response is central to fight pathogenic stimuli, overlapping mechanisms in primarily autoimmune inflammatory or degenerative diseases contribute to their perpetuation and induction of chronic tissue damage. In MS, chronic smoldering inflammation leads to demyelination and axonal damage, accompanied by the loss of oligodendrocytes and their precursors, which severely impairs endogenous remyelination and promotes the development of irreversible neurological deficits [19]. In PD, α-synuclein associated neuroinflammation predominantly affects dopaminergic neurons of the substantia nigra, contributing to their progressive loss and circuit-specific dysfunctions [20], while in AD the accumulation of amyloid-β plaques and tau tangles is associated with synaptic dysfunction and loss of glutamatergic neurons [21]. Despite their specific initiating factors and subsequent clinical manifestations, these diseases are unified by a chronically activated proinflammatory microglial response, leading to comparable outcomes including neuronal and glial cell death, axonal degeneration, synaptic loss, demyelination and function-hampering remodeling of the surrounding matrix [19–24]. Thus, inflammation-driven tissue damage represents a disease-overarching pathological factor operational in neuroinflammatory and -degenerative disorders.
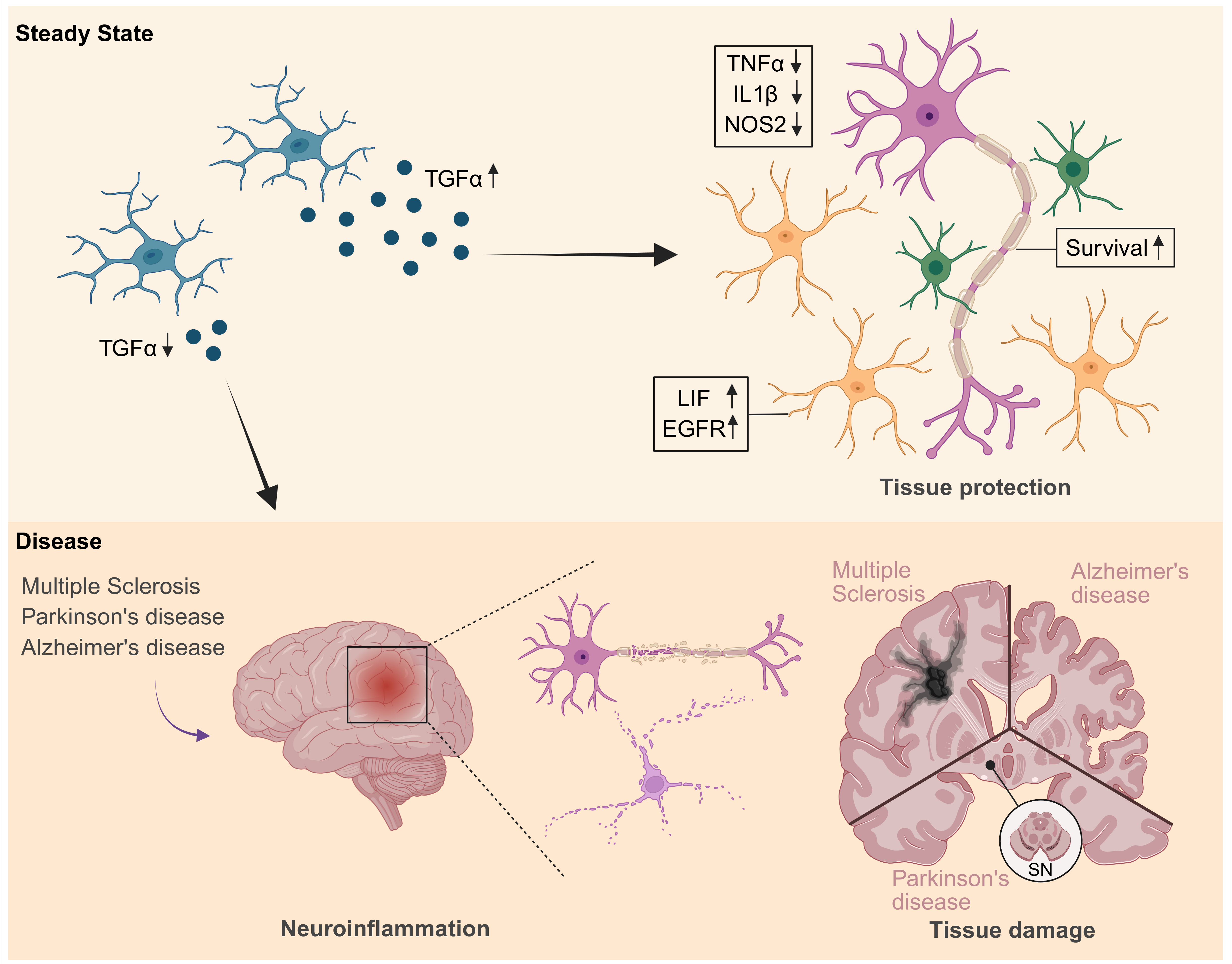
Figure 1. Graphical summary of TGFα mediated tissue protection during neuroinflammation. Neuroinflammatory diseases such as Multiple Sclerosis, Parkinson’s and Alzheimer’s disease are characterized by chronic inflammation in the central nervous system, leading to severe tissue damage. The production and release of the protective factor TGFα by microglia is proposed to contribute to limiting this inflammation-associated tissue damage. TGFα may act via EGFR signaling on neighboring cells to support tissue integrity and cell survival, thereby illustrating a potential CNS-intrinsic protective mechanism during neuroinflammation. SN: Substantia Nigra; Color code: Microglia (blue), neurons (pink), astrocytes (orange), oligodendrocytes (green). Created in BioRender. Ramphal, N. (2026) https://BioRender.com/931bzuy.
In this light, mechanisms that limit inflammatory responses, opposing pro-inflammatory degenerative cues and contributing to the restoration of tissue homeostasis are increasingly coming into focus. Indeed, anti-inflammatory and tissue-regenerative mediators represent a crucial regulatory mechanism. We and others have described several mechanisms of tissue-preservation and -recovery during autoimmune and degenerative CNS disorders [25–27], among those the transforming growth factor-α (TGFα), which is considered an important modulator of glial activation states, as its actions are capable to limit tissue-damage in the CNS.
TGFα is a member of the epidermal growth factor family and is synthesized as a transmembrane precursor, which is then secreted after cleavage by proteolytic shedding [28]. TGFα primarily signals through the epidermal growth factor receptor (EGFR) and activates MAPK/ERK, PI3K-Akt, PLCy/PKC and STAT signaling, thereby regulating various cellular programs including proliferation, survival, differentiation, migration and apoptosis [29–31]. This factor is particularly interesting as it has a context-dependent duality. In the peripheral system, TGFα is associated with pathological processes such as fibrosis or tumors [32,33]. However, in the CNS TGFα acts as a damage-modulating and protective signal rather than a classic growth factor. Here, TGFα is expressed by multiple cell types including neurons and glial cells [34,35]. Nevertheless, a recent study by Lößlein et al. [36] demonstrated that microglia represent the primary producers, an observation also supported by others [37].
It has been shown previously that TGFα is upregulated after CNS injuries, such as stroke or spinal cord trauma, and displays neuroprotective functions and regenerative activities [38,39]. These features raise the possibility that TGFα-EGFR signaling functions not only as a classical immunosuppressive pathway but as a tissue-intrinsic modulator that constrains inflammatory and potentially degenerative tissue damage. This concept is supported by the work of Lößlein et al., which provides evidence that TGFα provokes such damage-limiting signals.
The same study reveals the intriguing finding that TGFα exhibits precise temporal dynamics. Baseline levels are very low, but TGFα is upregulated during the peak phase of experimental autoimmune encephalomyelitis (EAE), the mouse model for MS [40], and subsequently normalizes again during recovery. This pattern suggests that the induction of TGFα coincides with a critical transition phase during which inflammatory responses trigger control mechanisms to protect the CNS from perceived danger and induce tissue-preserving and regenerative mechanisms. Parallel to TGFα expression, there is also dynamic EGFR upregulation in multiple cell types. This coordinated regulation of ligand and receptor suggests that TGFα-EGFR signaling is an endogenous CNS tissue-wide regulatory program for inflammation resolution rather than a cell type specific signaling pathway only. For instance, the resolution of inflammation is particularly relevant in MS lesions, which cause severe damage to CNS tissue. Previous work has already shown that elevated TGFα is essential for MS lesion resolution [37]. Consistent with this, Lößlein et al. demonstrate that microglial TGFα knockout leads to persistent inflammation, demyelination, axonal damage and neuronal death, establishing TGFα as a tissue-protective and regenerative factor.
A key conceptual advance of this work lies in demonstrating that TGFα exerts pleiotropic effects across multiple CNS and immune cell populations. Specifically, TGFα dampens the expression of pro-inflammatory mediators such as TNFα, IL1β and NOS2 in astrocytes, macrophages, dendritic cells and oligodendrocytes, while simultaneously promoting a protective astrocytic program characterized by the upregulations of leukemia inhibitory factor (LIF), another potent tissue-protective factor. Moreover, neurons also directly benefit from TGFα signaling, exhibiting enhanced survival and increased resistance to inflammatory and toxic insults. Rather than acting through a single effector pathway, TGFα coordinates a multicellular response that converges on a common outcome: the limitation of inflammatory tissue damage. Given the cellular complexity of the inflamed CNS, this pleiotropy constitutes a functional advantage for effective damage control.
In addition to these mechanistic observations, the work of Lößlein et al. also provides a therapeutic approach for neuroinflammatory diseases. The majority of currently available therapies, particularly in the field of MS, primarily target the modulation of peripheral immune activation or the migration of immune cells over the blood brain barrier (BBB). Even though these strategies are highly effective in reducing acute inflammatory events, they neither sufficiently address CNS-intrinsic mechanisms that determine the local tissue response induced by inflammatory cues, nor influence the balance between inflammatory tissue degeneration, chronic immune cell activation and irreversible tissue damage. The discovery that TGFα is a microglial signal that effectively limits inflammatory tissue damage therefore opens the possibility of novel therapeutic approaches: the targeted enhancement of the body’s own protective mechanisms within the inflamed CNS. For this, the study addressed one of the key translational barriers, which is limited BBB permeability of peripherally applied substances into the CNS. In a mouse model, intranasal administration of TGFα not only reduced the number of CNS infiltrating immune cells, but also decreased inflammatory tissue damage in both presymptomatic and symptomatic stages of EAE. This strategy provides compelling proof-of-concept for modulating inflammatory pathways in the CNS. The versatile properties of TGFα highlight the potential future avenue of carefully evaluating dose control, safety and long-term effects in translational contexts; as such, application of TGFα during limited time windows, e. g. during inflammatory relapses, may limit, among others, the potential harms of a proliferation inducing and tissue-remodeling agent. Despite these open challenges, these observations underscore TGFα as a systemic modulator of the inflammatory cascade and the associated damage. Finally, human data from Lößlein et al. contribute to the conceptual framework. The reduction of TGFα in the cerebrospinal fluid of MS patients and the associated clinical disability level (EDSS) suggest that insufficient TGFα-EGFR signaling is associated with increased tissue damage. However, the apparent conflict between elevated TGFα expression in inflamed CNS tissue [37] and reduced levels detected in the CSF points to a complex, maybe even compartment-specific regulation of this pathway. Such spatial separation could be particularly relevant for understanding how local deficits of TGFα or EGFR lead to tissue damage. Indeed, reduced EGFR signaling and related symptoms have already been observed in neurodegenerative diseases such as AD and PD [41], strengthening the notion that impaired EGFR signaling may represent a common feature across neuroinflammatory conditions.
The TGFα-EGFR signaling cascade may thus provide cross-disease protective function in CNS. The recurring association of reduced EGFR signaling with neurodegenerative and neuroinflammatory diseases suggest that TGFα may hold therapeutic potential not only for MS, but also for other CNS pathologies. Yet, it remains an unresolved issue to what extent TGFα mediated effects interact with other immunomodulatory signaling pathways or influence specific immune cell subpopulations involved in disease resolution, while the combination of immunomodulatory and tissue-protective effects continues to highlight TGFα as a promising candidate for innovative therapeutic strategies across diseases.
Together, the study by Lößlein et al., opens up a conceptual perspective for systematically investigating TGFα-based interventions. Addressing the open questions in future preclinical and clinical studies will be essential to fully define the therapeutic window of TGFα based strategies and to determine their broader applicability across neuroinflammatory and neurodegenerative disease contexts.
References
2. Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, et al. Neuroinflammation Induces Neurodegeneration. J Neurol Neurosurg Spine. 2016;1(1):1003.
3. Zindler E, Zipp F. Neuronal injury in chronic CNS inflammation. Best Pract Res Clin Anaesthesiol. 2010 Dec;24(4):551–62.
4. Harry GJ, Kraft AD. Neuroinflammation and microglia: considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab Toxicol. 2008 Oct;4(10):1265–77.
5. Hanamsagar R, Bilbo SD. Environment matters: microglia function and dysfunction in a changing world. Curr Opin Neurobiol. 2017 Dec;47:146–55.
6. von Bernhardi R. Glial cell dysregulation: a new perspective on Alzheimer disease. Neurotox Res. 2007 Dec;12(4):215–32.
7. Bohlen CJ, Friedman BA, Dejanovic B, Sheng M. Microglia in Brain Development, Homeostasis, and Neurodegeneration. Annu Rev Genet. 2019 Dec 3;53:263–88.
8. Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007 Nov;10(11):1387–94.
9. Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007 Mar 7;27(10):2596–605.
10. Jin WN, Shi SX, Li Z, Li M, Wood K, Gonzales RJ et al. Depletion of microglia exacerbates postischemic inflammation and brain injury. J Cereb Blood Flow Metab. 2017 Jun;37(6):2224–36.
11. Frei K, Fontana A. Antigen presentation in the CNS. Mol Psychiatry. 1997 Mar;2(2):96–8.
12. Shimizu T, Prinz M. Microglia across evolution: from conserved origins to functional divergence. Cell Mol Immunol. 2025 Dec;22(12):1533–48.
13. Prinz M, Masuda T, Wheeler MA, Quintana FJ. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu Rev Immunol. 2021 Apr 26;39:251–77.
14. Simard AR, Rivest S. Neuroprotective effects of resident microglia following acute brain injury. J Comp Neurol. 2007 Oct 20;504(6):716–29.
15. Rasley A, Tranguch SL, Rati DM, Marriott I. Murine glia express the immunosuppressive cytokine, interleukin-10, following exposure to Borrelia burgdorferi or Neisseria meningitidis. Glia. 2006 Apr 15;53(6):583–92.
16. Lindholm D, Castrén E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992 Apr;117(2):395–400.
17. Beal MF. Oxidative damage in neurodegenerative diseases. The Neuroscientist. 1997 Jan;3(1):21–7.
18. Muzio L, Viotti A, Martino G. Microglia in Neuroinflammation and Neurodegeneration: From Understanding to Therapy. Front Neurosci. 2021 Sep 24;15:742065.
19. Lassmann H, Brück W, Lucchinetti CF. The immunopathology of multiple sclerosis: an overview. Brain Pathol. 2007 Apr;17(2):210–8.
20. Morris HR, Spillantini MG, Sue CM, Williams-Gray CH. The pathogenesis of Parkinson's disease. Lancet. 2024 Jan 20;403(10423):293–304.
21. Abubakar MB, Sanusi KO, Ugusman A, Mohamed W, Kamal H, Ibrahim NH, et al. Alzheimer's Disease: An Update and Insights Into Pathophysiology. Front Aging Neurosci. 2022 Mar 30;14:742408.
22. Peterson JW, Bö L, Mörk S, Chang A, Trapp BD. Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol. 2001 Sep;50(3):389–400.
23. Herz J, Zipp F, Siffrin V. Neurodegeneration in autoimmune CNS inflammation. Exp Neurol. 2010 Sep;225(1):9–17.
24. Jang DG, Sim HJ, Song EK, Kwon T, Park TJ. Extracellular matrixes and neuroinflammation. BMB Rep. 2020 Nov;53(10):491–9.
25. Linnerbauer M, Lößlein L, Vandrey O, Peter A, Han Y, Tsaktanis T, et al. The astrocyte-produced growth factor HB-EGF limits autoimmune CNS pathology. Nat Immunol. 2024 Mar;25(3):432–47.
26. Güner F, Rothhammer V. Neurotrophic factors in multiple sclerosis. Front Immunol. 2025 Aug 27;16:1654603.
27. Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017 Jun 15;169(7):1276–90.e17.
28. Bosenberg MW, Pandiella A, Massagué J. The cytoplasmic carboxy-terminal amino acid specifies cleavage of membrane TGF alpha into soluble growth factor. Cell. 1992 Dec 24;71(7):1157–65.
29. Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004 Apr;286(4):L741–9.
30. Chalazonitis A, Kessler JA, Twardzik DR, Morrison RS. Transforming growth factor alpha, but not epidermal growth factor, promotes the survival of sensory neurons in vitro. J Neurosci. 1992 Feb;12(2):583–94.
31. Yarden Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer. 2001 Sep;37 Suppl 4:S3–8.
32. Korfhagen TR, Swantz RJ, Wert SE, McCarty JM, Kerlakian CB, Glasser SW, et al. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J Clin Invest. 1994 Apr;93(4):1691–9.
33. Calaf GM, Crispin LA, Muñoz JP, Aguayo F, Roy D, Narayan G. Ionizing Radiation and Estrogen Affecting Growth Factor Genes in an Experimental Breast Cancer Model. Int J Mol Sci. 2022 Nov 18;23(22):14284.
34. Tropepe V, Craig CG, Morshead CM, van der Kooy D. Transforming growth factor-alpha null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci. 1997 Oct 15;17(20):7850–9.
35. von Bossanyi P, Sallaba J, Dietzmann K, Warich-Kirches M, Kirches E. Correlation of TGF-alpha and EGF-receptor expression with proliferative activity in human astrocytic gliomas. Pathol Res Pract. 1998;194(3):141–7.
36. Lößlein L, Linnerbauer M, Zuber F, Tsaktanis T, Vandrey O, Peter A, et al. TGFα controls checkpoints in CNS resident and infiltrating immune cells to promote resolution of inflammation. Nat Commun. 2025 Jun 19;16(1):5344.
37. Rothhammer V, Borucki DM, Tjon EC, Takenaka MC, Chao CC, Ardura-Fabregat A, et al. Microglial control of astrocytes in response to microbial metabolites. Nature. 2018 May;557(7707):724–8.
38. Leker RR, Toth ZE, Shahar T, Cassiani-Ingoni R, Szalayova I, Key S, et al. Transforming growth factor alpha induces angiogenesis and neurogenesis following stroke. Neuroscience. 2009 Sep 29;163(1):233–43.
39. White RE, Yin FQ, Jakeman LB. TGF-alpha increases astrocyte invasion and promotes axonal growth into the lesion following spinal cord injury in mice. Exp Neurol. 2008 Nov;214(1):10–24.
40. Wekerle H. Experimental autoimmune encephalomyelitis as a model of immune-mediated CNS disease. Curr Opin Neurobiol. 1993 Oct;3(5):779–84.
41. Romano R, Bucci C. Role of EGFR in the Nervous System. Cells. 2020 Aug 12;9(8):1887.