Commentary
Current research investigating the pathomechanisms of neurodegenerative disorders of the central nervous system (CNS), such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), or Parkinson’s disease (PD), led to the understanding that these diseases have to be seen in the context of immune responses [1]. In other words, inflammation plays a central role in neurodegenerative disorders of the CNS. The ultimate question whether the immune responses that are observed in these diseases are causative or consequential, is debated. However, increasing evidence suggests that activation of both the adaptive and the innate immune system can drive the disease progression. One exemplary disease, in which both the adaptive and the innate immune system have been shown to be involved, is PD. The classical motor symptoms observed in PD patients are caused by the loss of dopaminergic (DA) neurons of the substantia nigra pars compacta (SNpc) [2-5]. DA neurons of the SNpc are thought to be particularly vulnerable because of their high levels of reactive dopamine, high energy demand and mitochondrial turnover, calcium handling, as well as their large axonal arborizations [6-8]. Although many causative gene mutations in familial forms have been analyzed, the underlying cause of sporadic PD is not understood. Interestingly, activated innate immune cells of the CNS, microglia, have been shown to directly contribute to the death of DA neurons in the midbrain [9]. Reactive microglia have been reported to be particularly abundant in the midbrain [10]. In addition, activated cells of the adaptive immune system, T lymphocytes, have also been observed in postmortem PD brain tissues [11]. These activated T cells seen in sporadic PD have furthermore been shown to actively kill DA neurons via the Interleukin 17 (IL-17) pathway. In summary, there is convincing evidence that immune response in the midbrain contributes directly to the loss of DA neurons in the SNpc. The biggest question that remains is: what causes this detrimental immune (over-)reaction? It has recently been reported that DA neurons are able to present alpha-synuclein as antigen causing activation of T cells. Why are T cells and microglia so abundant in the SNpc even before symptoms occur?
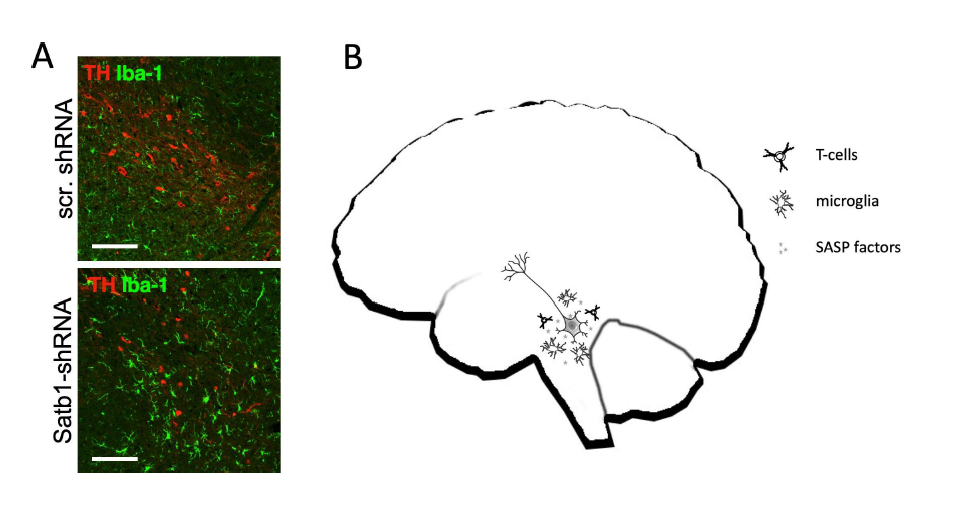
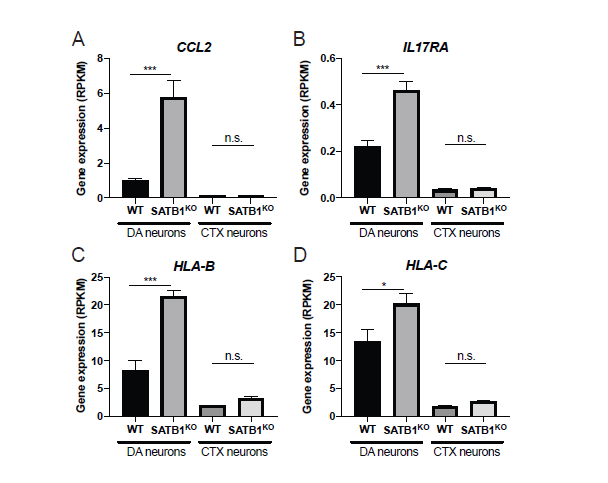
Recently, we discovered that post-mitotic DA neurons can become senescent. We have identified Special AT-Rich Sequence-Binding Protein 1 (SATB1) as a genetic master regulator that plays a neuroprotective role specifically in DA neurons of the SNpc [12]. Importantly, SATB1 was identified as a genetic risk factor for PD [13,14]. In summary, we sought to characterize the function of this transcriptional repressor in human DA neurons. When we knocked out SATB1 in pure cultures of human embryonic stem cell-derived DA neurons, we found that these cells first differentiate normally, but when they reach a mature state in which autonomous pacemaker activity starts, they become senescent [15]. Interestingly, stem cell-derived cortical (CTX) neurons were not affected by the loss of SATB1 and no differences between knockout (KO) and wild-type (WT) were observed. The senescence phenotype that was observed in both human SATB1-KO as well as mouse Satb1-knockdown DA neurons resembled every aspect of the phenotype typically described in mitotic cells. In the SATB1-KO DA neurons, we observed classical senescence hallmarks such as dysfunctional and swollen mitochondria, lysosomal dysfunction, elevated levels of reactive oxygen species (ROS) as well as oxidized proteins, lipofuscin accumulation, senescence associated β-galactosidase (SA β-gal) activity, increase of nuclear diameter in correlation with decreased lamin B1 expression, and activation of the senescence associated secretory phenotype (SASP). The SASP is characterized by the release of pro-inflammatory factors that cause a local inflammation in the surrounding tissue and recruits and activates immune cells detecting and removing senescent cells [16]. This mechanism is central to the immune response to senescent cells. Interestingly, we observed that senescent DA neurons in pure cultures persist, whereas senescent DA neurons in vivo trigger immune response and recruit microglia that actively remove the senescent DA neurons (Figure 1A) [15]. It is likely that the SASP is also attracting and activating T cells which could potentially kill senescent DA neurons. Re-assessment of the expression profile changes of the human ESC-derived DA neurons revealed a highly significant upregulation of the cytokine CCL2, the interleukin receptor IL17-R, and some major histocompatibility complex genes (MHC) such as HLA-B and HLA-C. It has previously been reported that CCL2 attracts T cells [17]. Moreover, it was proposed that T cells can actively kill DA neurons in PD patients via the IL17 pathway - specifically DA neurons which express IL17- R [18]. Our findings therefore implicate that senescent DA neurons actively trigger an immune response and attract immune cells such as T cells and microglia. Moreover, upregulation of receptors, such as IL17-R, actively mediate the removal of these cells. Given that alpha-synuclein accumulation has been shown to trigger a DNA damage response [19], it is very likely that there is an age-related increase in the number of DA neurons that express a program of cellular senescence which is induced by the activation of the DNA damage response. Occurrence of senescent cells in the SNpc would trigger a local inflammation and attract and activate both innate and adaptive immune cells which would actively remove the senescent DA neurons. This theory is in line with the finding of unusually high numbers of both microglia as well as T cells present in the SNpc of PD patients. This increase of immune cells occurs before the onset of symptoms of PD [20]. Even in healthy aging, there is a constant loss of nigral neurons throughout life, with an estimated ~10% loss in every decade of life starting around the age of twenty [21]. Since the DA neurons are particularly prone to senesce, we hypothesize that there is a constant turnover of cells that become senescent. These senescent cells are removed by an intact immune system. With age, it is thought that immune cells get more used to senescent cells and are not detected and removed as efficiently any more [22]. It is easily imaginable that if senescent cells remain in the midbrain, they will cause and sustain a local inflammation which can be detrimental for the present DA neurons. The SASP will trigger an inflammatory response and attract and activate immune cells (summarized in Figure 1A). Our RNA-Seq data from senescent human SATB1-KO DA neurons revealed a significant upregulation of SASP/immune genes that potentially attract immune cells. CCL2 was ~6-fold upregulated, but was not significantly altered in SATB1- KO CTX neurons (Figure 2A). CCL2 has been shown to attract T cells and could explain the high abundance of these immune cells in the SNpc of PD patients. Once in close proximity, T cells have been shown to kill DA neurons via the IL17 pathway. Interestingly, senescent DA neurons express significantly upregulated levels of IL17R, a gene that mediates neuronal cell death by IL-17– IL-17R signaling and activation of Nuclear Factor Kappa B (NFκB) (Figure 2B) [18]. In line with the discovery that antigen presentation of DA neurons is critical for the immune response [23], we found significantly elevated expression of Major Histocompatibility Complex (MHC) class I genes such as HLA-B and HLA-C which were not significantly changed in CTX SATB1-KO neurons (Figure 2C and 2D). In summary, there is striking evidence that DA neurons are particularly prone to senesce and that the presence of the SASP in the SNpc could potentially explain the occurrence of a local inflammation which in turn triggers a massive immune response and finally a removal of DA neurons. Importantly, we have previously demonstrated that senescent DA neurons react to senolytics treatment and can be killed by this new class of compounds. Given that senescent DA neurons seem to occur sporadically, it would potentially be feasible to interfere with a senolytics treatment to ameliorate the local inflammation and most importantly, to prevent spreading of senescence which could lead to dramatic loss of DA neurons and consequently to PD.
Acknowledgements
This work was supported by the United States Army Medical Research and Materiel Command (USAMRMC) under awards W81XWH-17-1-0495 and W81XWH-18-1-0467, and by NYSTEM contract C32598GG. I am grateful to D. Poulter and S. Stelter for commenting on the manuscript.
References
2. Bernheimer H, Birkmayer W, Hornykiewicz O, Jellinger K, Seitelberger F. Brain dopamine and the syndromes of Parkinson and Huntington Clinical, morphological and neurochemical correlations. Journal of the Neurological Sciences. 1973 Dec 1;20(4):415-55.
3. De Rijk MD, Launer LJ, Berger K, Breteler MM, Dartigues JF, Baldereschi M, et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology. 2000 Jan 1;54(11 Suppl 5):S21-3.
4. Fearnley JM, Lees AJ. Ageing and Parkinson’s disease: substantia nigra regional selectivity. Brain. 1991 Oct 1;114(5):2283-301.
5. Strickland D, Bertoni JM. Parkinson’s prevalence estimated by a state registry. Movement disorders: official journal of the Movement Disorder Society. 2004 Mar;19(3):318-23.
6. Riessland M, Kolisnyk B, Greengard P. Reactive dopamine leads to triple trouble in nigral neurons. Biochemistry 2017, 56(49):6409-6410.
7. Pacelli C, Giguère N, Bourque MJ, Lévesque M, Slack RS, Trudeau LÉ. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Current Biology. 2015 Sep 21;25(18):2349-60.
8. Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ- 1. Nature. 2010 Dec;468(7324):696-700.
9. Qian L, Flood PM. Microglial cells and Parkinson’s disease. Immunologic Research. 2008 Jul 1;41(3):155.
10. Kim WG, Mohney RP, Wilson B, Jeohn GH, Liu B, Hong JS. Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: role of microglia. Journal of Neuroscience. 2000 Aug 15;20(16):6309-16.
11. Garretti F, Agalliu D, Lindestam Arlehamn CS, Sette A, Sulzer D. Autoimmunity in Parkinson’s Disease: the role of a-synuclein-specific T cells. Frontiers in Immunology. 2019 Feb 25;10:303.
12. Brichta L, Shin W, Jackson-Lewis V, Blesa J, Yap EL, Walker Z, et al. Identification of neurodegenerative factors using translatome–regulatory network analysis. Nature Neuroscience. 2015 Sep;18(9):1325-33.
13. Hu X, Mao C, Hu Z, Zhang Z, Zhang S, Yang Z, et al. Association Analysis of 15 GWAS-linked Loci with Parkinson’s Disease in Chinese Han Population. Neuroscience Letters. 2020 Mar 9:134867.
14. Nalls MA, Blauwendraat C, Vallerga CL, Heilbron K, Bandres-Ciga S, Chang D, et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. The Lancet Neurology. 2019 Dec 1;18(12):1091- 102.
15. Riessland M, Kolisnyk B, Kim TW, Cheng J, Ni J, Pearson JA, et al. Loss of SATB1 induces p21-dependent cellular senescence in post-mitotic dopaminergic neurons. Cell Stem Cell. 2019 Oct 3;25(4):514-30.
16. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annual Review of Pathology: Mechanisms of Disease. 2010 Feb 28;5:99-118.
17. Berencsi K, Rani P, Zhang T, Gross L, Mastrangelo M, Meropol NJ, et al. In vitro migration of cytotoxic T lymphocyte derived from a colon carcinoma patient is dependent on CCL2 and CCR2. Journal of Translational Medicine. 2011 Dec 1;9(1):33.
18. Sommer A, Marxreiter F, Krach F, Fadler T, Grosch J, Maroni M, et al. Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell. 2018 Jul 5;23(1):123-31.
19. Milanese C, Cerri S, Ulusoy A, Gornati SV, Plat A, Gabriels S, et al. Activation of the DNA damage response in vivo in synucleinopathy models of Parkinson’s disease. Cell Death & Disease. 2018 Jul 26;9(8):1-2.
20. Galiano-Landeira J, Torra A, Pariente C, Vila M, Bové J. Immunogenic CD8+ T cell- mediated neuronal death as an initiator and progressor of Parkinson’s disease: A histological study of T cell subsets including iLBD postmortem tissue. In: SfN Meeting, 2019, Chicago. Chicago: 2019 Neuroscience Meeting Planner. Chicago, IL: Society for Neuroscience, 2019.
21. Reeve A, Simcox E, Turnbull D. Ageing and Parkinson’s disease: why is advancing age the biggest risk factor?. Ageing research reviews. 2014 Mar 1;14:19-30.
22. Van Deursen JM. The role of senescent cells in ageing. Nature. 2014 May;509(7501):439-46.
23. Cebrián C, Zucca FA, Mauri P, Steinbeck JA, Studer L, Scherzer CR, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nature communications. 2014 Apr 16;5(1):1- 4.