Keywords
Ovarian cancer, Immunotherapy, IL-2, Regulatory T cell, IL-2 receptor
IL-2 and IL-2 Receptor
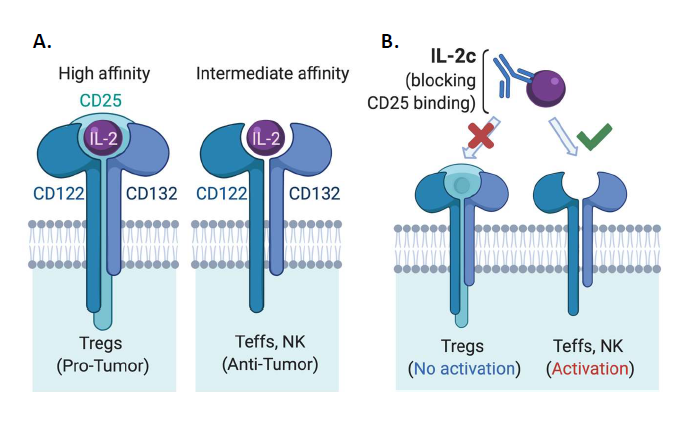
Interleukin (IL)-2 was first identified as a potent T cell growth factor in 1976 [1] and cloned in 1983 [2]. In the late 1990s, IL-2 gained further attention as the first immunotherapy demonstrating clinical efficacy against metastatic cancer, as high dose IL-2 was approved by the United States Food & Drug Administration (FDA) for the treatment of metastatic renal cell carcinoma in 1992 and metastatic melanoma in 1998 [3]. However, initial IL-2 immunotherapy treatment regimens led to severe and sometimes fatal toxicity in patients [4], significantly limiting its clinical application. Early treatment regimens were thought to require high doses of IL-2 for several reasons. IL-2 has a short in vivo half-life [5], requiring frequent dosing to maintain its biological effect of stimulating antitumor immunity. There are different forms of the IL-2 receptor (IL-2R) with different affinity for IL-2 [6] that express distinct cellular expression patterns (Figure 1A). Effector T cells (Teffs) and natural killer (NK) cells, linchpins of antitumor immunity, constitutively express intermediate-affinity IL-2Rs at baseline, which are dimeric receptors comprised of IL-2Rβ (CD122) and IL-2Rγ (common γ-chain or CD132) [6] subunits. Yet, a major immunosuppressive population in the tumor microenvironment (TME), regulatory T cells (Tregs) among other populations, express trimeric high-affinity IL-2Rs [6], which consist of IL-2Rβ, IL-2Rγ and IL-2Rα (CD25) subunits (Figure 1A). Therefore, unaltered IL-2 must be used at high doses for optimal Teff activation via intermediate affinity IL- 2Rs as a trimeric IL-2R binds IL-2 with roughly 10-100-fold higher affinity than a dimeric IL-2R [5], rendering Tregs a great competitive advantage for IL-2 availability over Teffs in the TME.
IL-2/anti-IL-2 mAb Complexes
To overcome limitations of native IL-2 in cancer treatment, many engineered forms or complexes of IL-2 have been developed in the past decade [7-21]. These approaches include PEGylated IL-2 agonists [18,19,22,23], IL-2 fusion proteins [12,23], IL-2 and anti-IL2 mAb complexes [7,24- 26], IL-2 with the CD25 binding site genetically deleted [27] and IL-2 fused to CD25 [28] among others [23,29], aiming preferentially activate antitumor effector cells over Tregs, and in some cases also to prolong IL-2 in vivo half-life. These agents are in different stages of preclinical/clinical research, recently reviewed elsewhere [23]. In particular, several groups have designed IL-2 agents that preferentially signal to Teffs through intermediate-affinity IL-2Rs over Tregs, including Boyman and colleagues who described selective activation of immune populations by distinct IL-2/anti-IL-2 immune complexes in 2006 [7]. This group used an anti-IL-2 mAb clone JES6-5H4 (S4B6-like) that could sterically block the interaction between IL-2 and CD25 while conformationally stabilizing the IL-2:CD122 interaction, leading to preferential activation of CD122 expressing (in the intermediate affinity IL-2R) Teffs and NK cells over Tregs [16] (Figure 1B). This CD122-selective IL-2 complex (IL-2c) is the focus of this commentary as several groups have previously shown the efficacy of IL-2c in multiple murine cancer models including B16/F10 melanoma [9,10,30], pancreatic cancer model [8], BCL1 leukemia [9], TRAMP-C1 sarcoma [14], and orthotopic bladder cancer [31].
IL-2c Treats an Immune Checkpoint Blockade Resistant Mouse Ovarian Cancer Model
Recently, we reported beneficial treatment effects of IL- 2c in a mouse ovarian cancer model [32] (major findings summarized in Figure 2), a significant advance in ovarian cancer (OC) immunotherapy for several reasons. OC is the deadliest gynecological cancer, leading to an estimated 13,700 deaths in the United States in 2021 [33]. It should respond to immunotherapies since intratumoral T cell infiltration predicts improved OC patient survival [34] which we showed is reduced by intratumoral Tregs [35]. However, decades of OC immunotherapy trials done by outstanding groups have been largely disappointing [36], which could be in part due to OC’s relatively low mutation burden compared to other tumors [36].
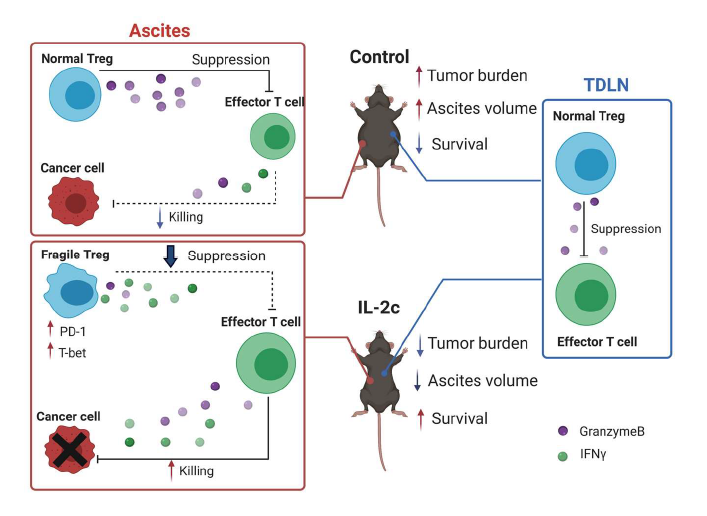
Figure 2. Graphical abstract of Drerup, Deng et al, Cancer Res. 2020 Nov 15;80(22):5063-5075. Major findings: 1) IL-2c inhibits growth of ID8agg mouse ovarian cancer and extends survival. 2) IL-2c increases effector T cell production of cytokines (e.g., IFNγ) and effector molecule (e.g., GranzymeB) in ascites.3) IL-2c decreases Treg production of effector molecule (e.g, GranzymeB) and inhibits Treg suppression in ascites, but not tumor draining lymph nodes. 4) IL-2c induces a fragile Treg phenotype in ascites, but not tumor draining lymph nodes. IL-2c synergizes with αPD-L1.
We found IL-2c is highly efficacious in the aggressive and poorly-responsive ID8agg mouse ovarian cancer line that we developed, including being refractory to immune checkpoint blockade (ICB) with α-programmed death ligand 1 (PD-L1). Consistent with known IL-2c effects, IL-2c promoted the activation of effector CD8+ T cells and CD4+ non-Treg cells and increased production of antitumor cytokines including IFNγ and TNFα by these cells in ID8agg challenge. Surprisingly however, IL-2c also expanded the number of Tregs thus reducing the CD8+ T cell/Treg ratio specially in ascites, an effect not previously reported with this approach, which could reflect the OC TME or OC cells themselves. Further investigation revealed that IL-2c treatment impaired Treg suppressive function in a TME-specific manner as Tregs from tumor draining lymph nodes (TDLN) suppressed normally.
Although the half-life of IL-2c is around 24 hours in vivo [11], we observed significant tumor-inhibitory effects as long as three weeks after the last dose of IL-2c. These data suggest induction of immune memory, evident by observed increases in central and effector memory CD8+ T cells after IL-2c treatment. Because IL-2c can increase TILs and tumors with pre-existing T-cell infiltration respond well to ICB [37], we explored combination of IL-2c and αPD-L1 therapy and found a synergistic effect of IL-2c plus αPD-L1 that was superior to either agent alone, in both (no t)ID8agg OC and B16 melanoma model. Moreover, mice that recovered from primary ID8agg challenge after combination therapy were resistant to subsequent tumor challenge, confirming induction of protective immune memory. Thus, IL-2c can work through simultaneous targeting of distinct immune pathways with effects that include: 1) activating effector T cells and promoting their antitumor cytokine production, 2) inhibiting immune-suppressive Tregs, and 3) inducing durable, protective immune memory.
In recent work, we performed additional exploration into the specific immune population(s) responsible for IL-2c efficacy in ID8agg tumors. CD8+ T cells are the primary target populations of IL-2c and are required for successful IL-2c treatment in several tumor models [9,14]. However, we found IL-2c was efficacious in ID8agg tumors following antibody-mediated CD8+ T cell depletion in vivo depletion. We tested two depletion timepoints, during the course of IL-2c treatment starting in week 2 or a week post the last dose of IL-2c (week 3), both of which significantly depleted CD8+ TILs for the duration of the experiment (Figure 3B) and did not affect IL-2c efficacy (Figure 3A). This result was striking because despite IL-2c mediated activation of CD8+ TILs in other models (Table 1), they were dispensable for IL-2c efficacy here. We also recently published that in orthotopic MB49 mouse bladder cancer, CD8 T+ cells were not required for IL-2c efficacy, which instead relied on δ T cells [31]. However, IL-2c treated ID8agg tumors effectively in γδ T cell-deficient mice [31], consistent with TME-specific IL-2c effects.
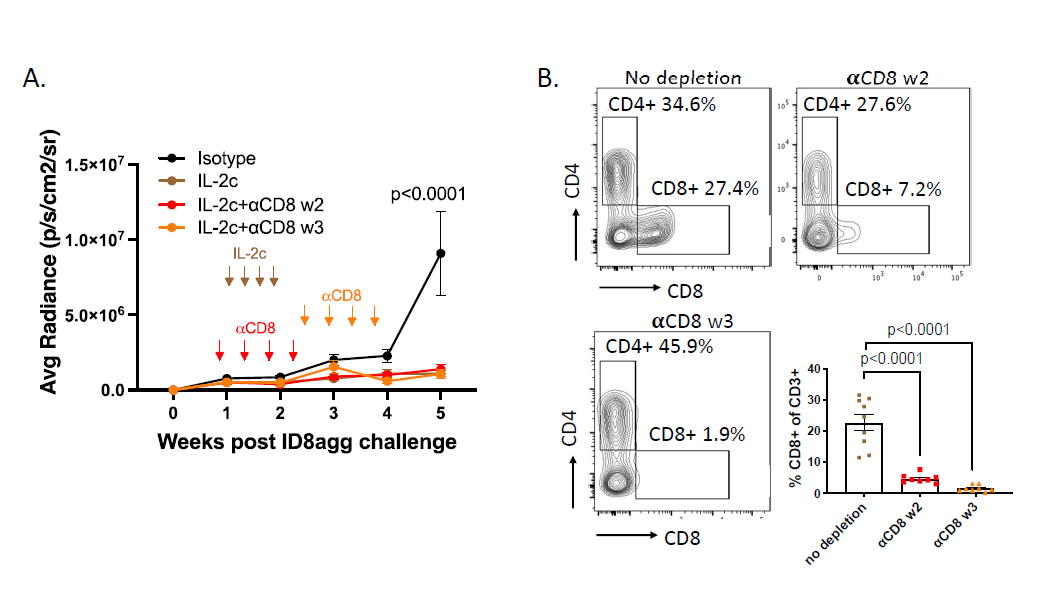
Figure 3. CD8+ conventional T cells are not required for the treatment efficacy of IL-2c. A. ID8agg tumors measured by luciferase as described [30]. Arrows indicate treatment of IL-2c or CD8 depleting antibody. N=8-9 per group, p value determined by two-way ANOVA. B. Flow analysis of tumor harvested on day 36 post ID8agg challenge. Contour plots are derived from CD3+ TILs. P value determined by one-way ANOVA.
| Author | Year | Agent Tested | Immune cell activation invitro | Immune cell activation in non- tumor bearing mice | in vivo efficacy | Combination therapy | Population responsiblefor efficacy | Immune change in TME | Ref |
|---|---|---|---|---|---|---|---|---|---|
| Boyman et al | 2006 | IL-2/mAb complex | Mouse & human Teff proliferation | 7 | |||||
| Kamimura et al | 2006 | antiIL-2 mAb + IL-2 cDNA | Expansion of CD8, NKcells, decrease Tregs | B16 lung metastasis | 24 | ||||
| Verdeil etal | 2008 | IL-2/mAb complex | Mouse & human Teff proliferation, cytokine production | pancreatic model | vaccination | CD8 expansion, pSTAT5, effector molecule cytokine production | 8 | ||
| Jin et al | 2008 | IL-2/mAb complex | Expansion of DC, NK, CD8 in spleen, increased perforin and GB for NK cells | B16 lung metastasis | NK cells required, but not DCs | 30 | |||
| Tomala etal | 2009 | IL-2/mAb complex | Both S4B6 and JES6-1 are able to activate CD8 T cells; Only S4B6 activate NK cells | B16 SQ | CD8 and NK cells required | 9 | |||
| BCL1 leukemia | CD8 required | ||||||||
| Letourneau et al | 2010 | IL-2/mAb complex | CD8+ t cell proliferation | 11 | |||||
| Krieg et al | 2010 | IL-2/mAb complex | Expansion of CD8, NKand Tregs | B16 SQ, B16 lung |
10 | ||||
| Redmond et al | 2012 | IL-2/mAb complex | MCA-205 sarcoma |
OX40 agonistAb |
T cells required | 14 | |||
| TRAMP-C1- mOVA prostate |
[doesn't respond to IL-2c alone] | Expansion of OT-1 CD8 (ki67,GB, KLRG1) | |||||||
| Cho et al | 2012 | IL-2/mAb complex | B16 SQ | vaccination, adoptive T cell transfer, aPD-L1 |
21 | ||||
| Raeber etal | 2020 | IL-2/mAb complex | No cDC actiavation in vitro |
cDC expansion in vivo through IL-2 stimulated ILCs | B16 SQ, Braf melanoma | B16: DC is required | Expansion of cDC1 in the tumor | 25 | |
| Drerup et al | 2020 | IL-2/mAb complex | ID8agg OC,B16 SQ | aPD-L1 | Activation of tumor CD8+ and CD4+ non-Tregs with increased production of anti-tumor cytokines. Induce Tregfragility in local TME but not TDLN. |
32 | |||
| Reyes et al | 2021 | IL-2/mAb complex | MB49, MBT-2 orthotopic Bladder Cancer, MB49 metastatic lung |
aPD-L1 | MB49: CD8 not required; gdT cells required |
No activation of CD8+ seen inMB49 orthotopic. Significantly increased intratumoral NK and γδ T cellcontent, activation , and effector function |
31 | ||
| Gillies etal | 2011 | NHS-IL2LT IL-2 variant fusion with an antibody (NHS76) |
mouse CD8, CD4 (proliferation) | LLC lung cancer | CD8 and NK cells required, CD4 not required | 12 | |||
| NXS2 neuroblastoma liver metastasis |
|||||||||
| Levin et al | 2012 | IL-2 superkine & IL-2/mAb complex | human NK cell pSTAT5 status (also in CD25-/-) | CD8+ expansion | B16 SQ; B16 lung; MC38,Lewis lung carcinoma |
13 | |||
| Carmenate et al | 2013 | human IL-2 mutein | Mouse NK, CD8 proliferation but not Tregs | MB16F0 melanoma lung metastasis |
CD8 and NK cells required | 15 | |||
| 3LL-D122 mouse lungcarcinoma | |||||||||
| Charych et al | 2016 | Bempegaldesleu kin NKTR-214 (pegylated IL-2) |
mouse T cells pSTAT5 | B16 SQ | aCTLA-4 | Expansion of CD8, NK, decreased CD4, increased intratumoral CD8/Treg ratio | 22 | ||
| EMT6 breast cancer, CT26 colon model | |||||||||
| Charych et al | 2017 | Bempegaldesleu kin: NKTR-214 (pegylated IL-2) |
pSTAT5 on CD3+ in PB | MBT-2 (bladder), H22(liver), Pan02 (pancreatic) | 18 | ||||
| Sockolosky et al | 2018 | orthoIL2 | (CD8, Tregs)pSTAT5, proliferation |
B16 | Increased CD8IFNg production, PD-1 Tim3 expression | 17 | |||
| Sun et al | 2019 | Ab-sumIL2 | B16 | aPD-L1 | CD8 required; NK, MDSC, CD4 not required |
Increased CD8/Treg ratio, increased CXCR5+PD-1+ cells,increase PD-L1 on DCs | 40 | ||
| MC38 | |||||||||
| Sahin et al | 2020 | hIL-2/NARA1 | pSTAT5 comparison Cytotoxic T cells, NK cell, Tregs | pSTAT5, Ki67, cell counts, for CD8, NK and Tregs | B16 (sQ and metastatic) LLC lung; 4T1breast | 4T1: doesn't treat primary as single agent but reduces metastasis |
hIL-2/NARA1: increase CD8, NK1.1, Treg; NARA1leukin: not Treg; lower exhausted Tcells | 26 | |
| Sharma et al | 2020 | Bempegaldesleu kin | B16, Colon (CT- 26), ovarian (BR5FVB), bladder (MBT- 2), liver (H22), pancreatic (Pan02), breast (EMT-6), lung (LLC), |
aPD-1, vaccine |
19 | ||||
| Hsu et al | 2021 | ProIL2 | MC38, CT26,B16 | aPD-L1 | Increased CD8, decreased Foxp3, increased ratio | 29 |
IL-2c Induces TME Treg Fragility Independent of CD8+ and δ T cells
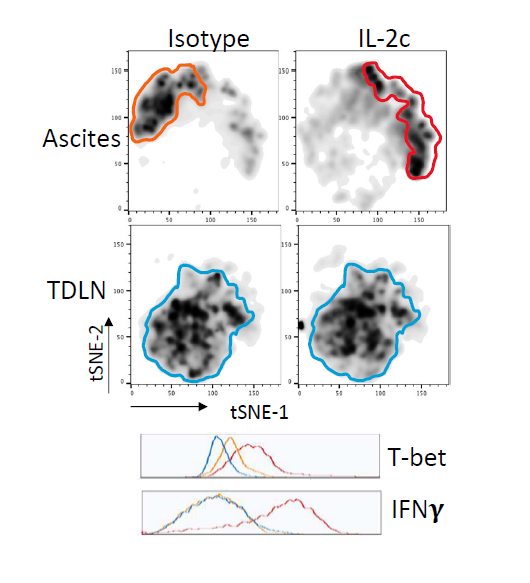
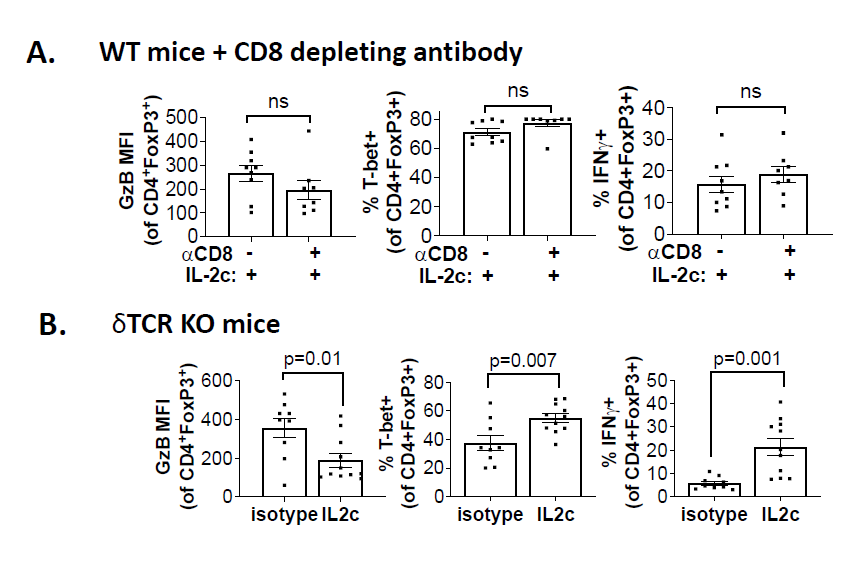
Tregs are a major immune-suppressive population in the TME and are associated with worse outcomes in OC patients [35]. Tregs also play a crucial role in the development of ID8agg tumors as we previously showed that depleting Tregs completely abolished ID8agg tumor progression [32]. Strikingly, fragile Tregs were first described by the Vignali group and are characterized by maintained FoxP3 expression with loss of suppressive function and decreased production of effector molecules including granzyme B, versus an unstable Treg phenotype where Tregs lose their FoxP3 expression [38]. Fragile Tregs also express transcription factor T-bet and produce cytokines that are uncharacteristic of normal Tregs [39], including IFNγ. Treg fragility can also contribute to ICB treatment efficacy [38,39] which could be another mechanism of IL-2c synergy with αPD-L1. We found that Tregs displayed a fragile phenotype induced by IL-2c, which helps explain IL-2c efficacy despite increasing Treg numbers in the OC TME. We performed t-distributed stochastic neighbor embedding (tSNE) analysis of Tregs in the OC TME and TDLN and found that Tregs in TDLN treated with control or IL-2c have a similar immune phenotype, whereas IL-2c-treated Tregs in the ascites acquired the fragile phenotype (Figure 4). By contrast, IL-2c did not induce Treg fragility in the bladder in MB49 bladder cancer, consistent with TME-specific fragility induction.
We further explored mechanisms for IL-2c-induced Treg fragility. We observed that IL-2c-induced fragility can be detected three weeks after the final dose of IL-2c, but not one week after the final dose [32], and is likely indirect from CD25 signals. Ex vivo treatment of Tregs with IL-2c does not fully recapitulate the in vivo fragile phenotype [32], suggesting IL-2c cooperates with another factor or population in the OC TME to induce Treg fragility, or could work during Treg differentiation, which could account for the long time needed for in vivo fragility induction. IFNγ is a potent Treg fragility inducer [38] and IL-2c promotes IFNγ production in effector T cells. We assessed major IFNγ-producing population effects on Treg fragility induction, including CD8+ T cells and γδ T cells. Antibody-mediated CD8+ T cell depletion did not rescue the Treg fragility phenotype in ascites (Figure 5A), and fragile Tregs with increased T-bet, and IFNγ expression were also present in ascites of γδ T cell deficient mice challenged with ID8agg tumors (Figure 5B) excluding δ T cells and making CD8+ T cells unlikely mediators in IL-2c induced Treg fragility. Future studies will test NK cell, innate lymphoid cell and fragile Treg interferon-γ in IL-2c induced Treg fragility and assess other tumors and TME.
Conclusions and Future Directions
Our published studies [31,32] and new data shown here highlight several important mechanisms that could be unique to the OC TME and serve as important contrasts to other studies using the same IL-2c or similar CD122-biased modified IL-2. First, conventional CD8+ T cells are not required for the treatment efficacy of IL-2c in ID8agg OC, despite IL- 2c-mediated promotion of IL-2 signaling and antitumor cytokines production in intratumoral CD8+ T cells. In this specific TME, IL-2c preferentially activates other immune populations that play a more crucial role than CD8+ T cells in treatment efficacy. Absence of OC TME CD8+ T cells is not seen and thus agents such as αPD-L1 that do activate OC TME CD8+ T cells can synergize with IL-2c as we reported. Other IL-2c activated populations including NK cells or other members of the innate immunity could be preferentially activated over CD8+ T cells or insufficient CD8+ T cells could induce compensatory activation of other cell populations. Understanding the preferential and distinct IL-2 signaling and activation of varied immune populations in specific TMEs and/or tumors is essential for comparing differences in IL-2 targeting agent mechanisms between various tumors and anatomic locations, and to define mechanisms for agents that can cooperate clinically with directed IL-2 agents, which to date is little studied. Second, IL-2c inhibits intratumoral Treg function by inducing a fragile phenotype at least in some tumors and environments. CD122-selective IL-2 agents are expected to avoid activation and proliferation of Tregs and most previous studies in other models observed an increased CD8+ T cell/Treg ratio [8,14,22,40] and sometimes even Treg depletion [19]. However, we found increased number of Tregs and fragility in ascites after IL-2c treatment. Studies are warranted to identify fragility mediators, define in what other cancer models or human cancers fragile Tregs can be induced and to understand which agents best combine with this effect for treatment efficacy.
Currently, many modified IL-2 agents are in varied stages of preclinical and clinical development [23] (and Table 1), yet a detailed understanding of the immune landscape during treatment is lacking in most cases. Our prior and ongoing investigations have uncovered unexpected yet useful immune consequences of IL-2c signaling in the OC TME, stressing the importance of understanding the biology and immunology of distinct IL-2 agents to advance our understanding of IL-2 and IL-2 receptor biology, and optimal therapeutic uses. We also shed light on OC-specific TME phenotypes that potentially help explain its unresponsiveness to current immunotherapies, which should facilitate further design of targeted immunotherapies for OC patients.
Funding
YD (CPRIT Research Training Award (RP170345) and Ovarian Cancer Research Alliance Ann and Sol Schreiber Mentored Investigator Award), R. Reyes (NIH T32GM113896, NIH/NCATS TL1 TR002647, NIA T32 AG 021890), T. Curiel (CA054174, CA205965, CDMRP, The Owens Foundation, The Skinner endowment, The Barker endowment, P30 CA054174) and the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant UL1 TR002645.
Conflict of Interest Statement
All authors declared no conflict of interest.
References
2. Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983 Mar;302(5906):305-10.
3. Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. The Journal of Immunology. 2014 Jun 15;192(12):5451-8.
4. Majidpoor J, Mortezaee K. Interleukin-2 therapy of cancerclinical perspectives. International Immunopharmacology. 2021 Sep 1;98:107836.
5. Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: biology, design and application. Trends in Immunology. 2015 Dec 1;36(12):763-77.
6. Malek TR. The biology of interleukin-2. Annual Review of Immunology. 2008 Apr 23;26:453-79.
7. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science. 2006 Mar 31;311(5769):1924-7.
8. Verdeil G, Marquardt K, Surh CD, Sherman LA. Adjuvants targeting innate and adaptive immunity synergize to enhance tumor immunotherapy. Proceedings of the National Academy of Sciences. 2008 Oct 28;105(43):16683-8.
9. Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. The Journal of Immunology. 2009 Oct 15;183(8):4904-12.
10. Krieg C, Létourneau S, Pantaleo G, Boyman O. Improved IL-2 immunotherapy by selective stimulation of IL-2 receptors on lymphocytes and endothelial cells. Proceedings of the National Academy of Sciences. 2010 Jun 29;107(26):11906-11.
11. Létourneau S, van Leeuwen EM, Krieg C, Martin C, Pantaleo G, Sprent J, et al. IL-2/anti-IL-2 antibody complexes show strong biological activity by avoiding interaction with IL-2 receptor α subunit CD25. Proceedings of the National Academy of Sciences. 2010 Feb 2;107(5):2171-6.
12. Gillies SD, Lan Y, Hettmann T, Brunkhorst B, Sun Y, Mueller SO, et al. A low-toxicity IL-2–based immunocytokine retains antitumor activity despite its high degree of IL-2 receptor selectivity. Clinical Cancer Research. 2011 Jun 1;17(11):3673-85.
13. Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012 Apr;484(7395):529-33.
14. Redmond WL, Triplett T, Floyd K, Weinberg AD. Dual anti-OX40/ IL-2 therapy augments tumor immunotherapy via IL-2R-mediated regulation of OX40 expression. PloS One. 2012 Apr 4;7(4):e34467.
15. Carmenate T, Pacios A, Enamorado M, Moreno E, Garcia- Martínez K, Fuente D, et al. Human IL-2 mutein with higher antitumor efficacy than wild type IL-2. The Journal of Immunology. 2013 Jun 15;190(12):6230-8.
16. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to interleukin-2 elicit selective T cell subset potentiation through distinct conformational mechanisms. Immunity. 2015 May 19;42(5):815-25.
17. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018 Mar 2;359(6379):1037-42.
18. Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PloS One. 2017 Jul 5;12(7):e0179431.
19. Sharma M, Khong H, Fa’ak F, Bentebibel SE, Janssen LM, Chesson BC, Creasy CA, Forget MA, Kahn LM, Pazdrak B, Karki B. Bempegaldesleukin selectively depletes intratumoral Tregs and potentiates T cell-mediated cancer therapy. Nature Communications. 2020 Jan 31;11(1):1-1.
20. Quixabeira DC, Zafar S, Santos JM, Cervera-Carrascon V, Havunen R, Kudling TV, et al. Oncolytic Adenovirus Coding for a Variant Interleukin 2 (vIL-2) Cytokine Re-Programs the Tumor Microenvironment and Confers Enhanced Tumor Control. Frontiers in Immunology. 2021 May 18;12:1827.
21. Cho HI, Reyes-Vargas E, Delgado JC, Celis E. A potent vaccination strategy that circumvents lymphodepletion for effective antitumor adoptive T-cell therapy. Cancer Research. 2012 Apr 15;72(8):1986-95.
22. Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an engineered cytokine with biased IL2 receptor binding, increased tumor exposure, and marked efficacy in mouse tumor models. Clinical Cancer Research. 2016 Feb 1;22(3):680-90.
23. Overwijk WW, Tagliaferri MA, Zalevsky J. Engineering IL-2 to give new life to T cell immunotherapy. Annual Review of Medicine. 2021 Jan 27;72:281-311.
24. Kamimura D, Sawa Y, Sato M, Agung E, Hirano T, Murakami M. IL-2 in vivo activities and antitumor efficacy enhanced by an anti-IL-2 mAb. The Journal of Immunology. 2006 Jul 1;177(1):306-14.
25. Raeber ME, Rosalia RA, Schmid D, Karakus U, Boyman O. Interleukin-2 signals converge in a lymphoid–dendritic cell pathway that promotes anticancer immunity. Science Translational Medicine. 2020 Sep 16;12(561).
26. Sahin D, Arenas-Ramirez N, Rath M, Karakus U, Hümbelin M, van Gogh M, Borsig L, Boyman O. An IL-2-grafted antibody immunotherapy with potent efficacy against metastatic cancer. Nature Communications. 2020 Dec 22;11(1):1-2.
27. Klein C, Waldhauer I, Nicolini VG, Freimoser-Grundschober A, Nayak T, Vugts DJ, et al. Cergutuzumab amunaleukin (CEA-IL2v), a CEA-targeted IL-2 variant-based immunocytokine for combination cancer immunotherapy: Overcoming limitations of aldesleukin and conventional IL-2-based immunocytokines. Oncoimmunology. 2017 Mar 4;6(3):e1277306.
28. Lopes JE, Fisher JL, Flick HL, Wang C, Sun L, Ernstoff MS, et al. ALKS 4230: a novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. Journal for Immunotherapy of Cancer. 2020;8(1).
29. Hsu EJ, Cao X, Moon B, Bae J, Sun Z, Liu Z, et al. A cytokine receptor-masked IL2 prodrug selectively activates tumor-infiltrating lymphocytes for potent antitumor therapy. Nature Communications.2021 May 13;12(1):1-3.
30. Jin GH, Hirano T, Murakami M. Combination treatment with IL-2 and anti-IL-2 mAbs reduces tumor metastasis via NK cell activation. International Immunology. 2008 Jun 1;20(6):783-9.
31. Reyes RM, Deng Y, Zhang D, Ji N, Mukherjee N, Wheeler K, et al. CD122-directed interleukin-2 treatment mechanisms in bladder cancer differ from αPD-L1 and include tissue-selective γδ T cell activation. Journal for Immunotherapy of cancer. 2021;9(4).
32. Drerup JM, Deng Y, Pandeswara SL, Padrón ÁS, Reyes RM, Zhang X, et al. CD122-Selective IL2 complexes reduce immunosuppression, promote Treg fragility, and sensitize tumor response to PD-L1 blockade. Cancer Research. 2020 Nov 15;80(22):5063-75.
33. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA: A Cancer Journal for Clinicians. 2021 Jan;71(1):7-33.
34. Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proceedings of the National Academy of Sciences. 2005 Dec 20;102(51):18538-43.
35. Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nature Medicine. 2004 Sep;10(9):942-9.
36. Drerup JM, Liu Y, Padron AS, Murthy K, Hurez V, Zhang B, et al. Immunotherapy for ovarian cancer. Current Treatment Options in Oncology. 2015 Jan 1;16(1):1.
37. Pitt JM, Vétizou M, Daillère R, Roberti MP, Yamazaki T, Routy B, et al. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and-extrinsic factors. Immunity. 2016 Jun 21;44(6):1255-69.
38. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ drives Treg fragility to promote antitumor immunity. Cell. 2017 Jun 1;169(6):1130-41.
39. Overacre-Delgoffe AE, Vignali DA. Treg fragility: a prerequisite for effective antitumor immunity?. Cancer Immunology Research. 2018 Aug 1;6(8):882-887.
40. Sun Z, Ren Z, Yang K, Liu Z, Cao S, Deng S, et al. A next-generation tumor-targeting IL-2 preferentially promotes tumor-infiltrating CD8+ T-cell response and effective tumor control. Nature Communications. 2019 Aug 28;10(1):1-2.