Abstract
Novel virus Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is highly infectious, which often causes severe acute and long-term illness, resulting in an increased rate of mortality and morbidity. Despite human cells being equipped with an antiviral innate immune system, SARS-CoV-2 quickly spread worldwide. A number of Pattern-Recognition Receptors (PRRs) are employed by cells to detect coronavirus infection, and timely interferon responses is immensely effective against SARS-CoV-2. However, the virus is also able to disarm the cell, attacking interferon-associated signaling pathways on multiple levels. Here we systematically analyze the complicated interactions between SARS-CoV-2 proteins and the host interferon signaling system, highlighting the role of cytosolic events in COVID-19 pathogenesis, and summarize a few antiviral pharmaceutic candidates that are potentially able to reduce morbidity and mortality of COVID-19.
Introduction
Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2), the causative agent of COVID-19 respiratory disease, was first identified on 7 December 2019 [1], and quickly spread worldwide. The SARS-CoV-2 belongs to Coronaviridea family, enveloped single-stranded RNA viruses. The family is divided into two subfamilies, the Coronavirinae and the Torovirinae, distinguished by the shape of their nucleocapsids [2]. Virions are roughly spherical and are notable for the large spike (S) glycoprotein that mediate viral entry to the host cell. The subfamily Coronavirinae consists of four genera, the alpha-, beta-, gamma-, and delta-coronaviruses. There are seven coronaviruses that can infect people – four of them causing a common cold (229E, NL63, OC43, HKU1), and three are associated with potentially severe respiratory conditions, namely severe syndrome coronavirus (SARS-CoV), Middle East respiratory syndrome coronavirus (MERS-CoV), and the emerging type of SARS-CoV-2, which has 79% sequence homology with SARS-CoV [1].
Life Cycle of SARS-CoV-2
The factors that are responsible for unusually high pathogenicity of SARS-CoV and MERS-CoV are not completely understood. However, research on these pathogens laid the ground for quick deciphering of the SARS-CoV-2’s life cycle. Coronaviruses are the largest known RNA viruses, with genomes ranging from 25 kb to 32 kb and 118-140 nm in virions diameter. Almost two-thirds of the genome encodes nonstructural proteins (nsps), participating in transcription, RNA genome replication, and counter-immune activities. Coronavirus nsps are synthesized as long precursor polypeptides, cleaved by virally encoded proteases. Among these, nsp12 is the large RNA-dependent RNA polymerase (RdRp), which in complex with other CoV Nsps replicates RNA genome. Another part of the genome encodes structural genes S, M, N, E or HE (torovirus), corresponding to spike, membrane, nucleocapsid, and envelop proteins, respectively [2].
Like SARS-CoV, the SARS-CoV-2 harbors large positive-sense single-stranded RNA genome (ssRNA), encoding nsps, accessory proteins and four structural proteins: spike (S) protein, for viral entry; envelope (E) protein, forming a pivotal ion channel in viral maturation and propagation process; membrane (M) protein, for viral structure assembly and binding with nucleocapsid (N) protein, which binds to the viral RNA itself [1].
The receptor-binding domain (RBD) of SARS-CoV-2’s spike protein recognizes a receptor protein on the cell membrane called angiotensin-converting enzyme 2 (ACE2) [3-5]. The interaction between S protein and ACE2 is facilitated by neuropilin-1 [6]. Upon ACE2 binding, SARS-CoV-2 utilizes two strategies to get inside of the cell depending on the expression patterns of host cell surface proteinase [7,8]. On the one hand, the spike protein is cleaved by proteases, such as cathepsin-L, which leads to endocytosis of virus and the receptor. On the other hand, the spike protein of SARS-CoV-2 can also be cleaved by transmembrane protease serine 2/4 (TMPRSS2/4), which creates a hydrophobic pocket that can rapidly bury itself into the closest membrane. The spike protein then folds back onto itself, by which the viral genome can be ejected from the virus into the host cytoplasm directly [5,7-9]. Once inside the host cell, the viral genomic RNA functions as a messenger RNA (mRNA), which is subsequentially translated by the host ribosomes.
The SARS-CoV-2’s genomic RNA consists of at least 14 open reading frames (ORFs) (Figure 1). The first ORF accounts for two-thirds of its genome and there are reasons for it. This giant ORF1 is the first ORF that is read by host ribosomes and codes two big polyproteins, pp1a and pp1ab, which are subsequentially cut into 16 nonstructural proteins (nsp1–11 from pp1a, nsp1–10 and nsp12–16 from pp1ab) by the cysteine proteases 3-chymotrypsin–like proteases (3CLpro, nsp5) and papain-like proteases (PLpro, nsp3) [4,10]. After released, nsp1-16 form the replication-transcription complex that synthesize negative RNA template and translate viral mRNA [1]. Nsp2-16 form viral replication and transcription complex (RTC) that is involved in viral RNA synthesis, RNA proofreading and RNA modification. Similar to other coronaviruses, SARS-CoV-2’s genome RNA is synthesized by nsp12 RNA-dependent RNA polymerase (RdRp) with two cofactors, nsp7 and nsp8. The RdRp has been proved as a centerpiece in the virus life cycle both for replication of the viral genome and also for transcription of subgenomic RNAs (sgRNAs) [11]. Some of the nsps (nsp3, nsp4, nsp6) have transmembrane domains and they serve to anchor the replication–transcription complex to the cell endomembranes and turn them into replication organelles, a prerequisite for the synthesis of additional viral RNAs [12,13].
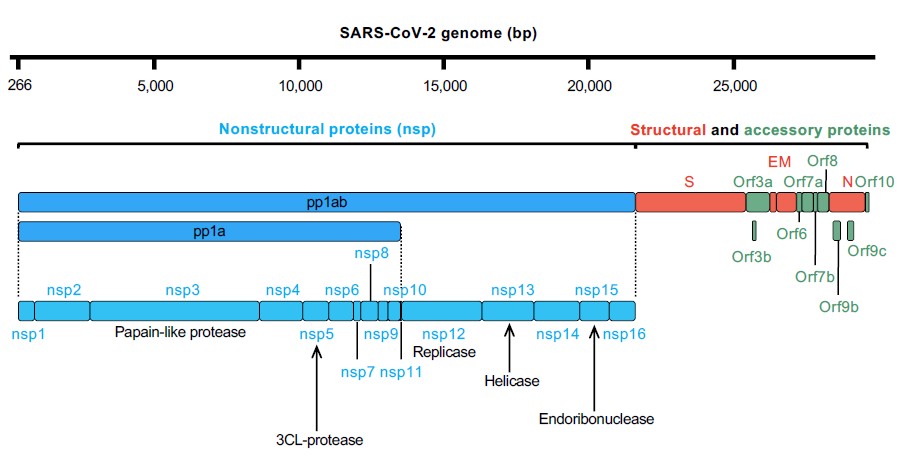
To successfully settle in the host, the virus must replicate itself as many as possible before it gets eliminated by the host immunity or loses genome integrity due to mutations. In the pathologic organelle termed ERGIC (endoplasmic reticulum-to-Golgi intermediate compartments), four essential structural proteins (S, E, M, N) together with the genomic ssRNA are assembling into complete virions, and subsequently encapsulated and released from the host cell for another round of infection [14].
Fortunately, the human cells developed multiple strategies during evolution to counter viral infection by activating interferon response, modulated by pattern-recognition receptors (PRRs), the main antiviral defense in all cells of the organism.
Pattern Recognition Receptors and Interferons
Pattern-recognition receptors
Pattern recognition receptors (PRRs) are intracellular molecular sensors recognizing abnormal and pathogen-associated molecular patterns (PAMPs), such as double-stranded RNA, unmethylated DNA, bacterial or viral fragments [15]. Since PAMPs are unique and highly conserved molecular structures associated with the specific kind of pathogenic microorganisms, host organisms evolutionally developed a set of PRR sensors and gene-responders, defined as the innate immune system [16-19]. Not only viral infection, but the host also produces some proteins and metabolites that PRRs could recognize products of cell necrosis, tissue damage, aberrant transcripts, or proteins. Such host molecules were defined as “damage-associated molecular patterns” (DAMPs) [18-20]. PPRs are expressed in immune cells, including monocytes, neutrophils, macrophages, dendritic cells, natural killer (NK) cells, mast cells, eosinophils, and basophils [21], and non-immune cells such as fibroblasts and epithelium cells [22]. PPRs can directly recognize specific PAMPs or DAMPs on the surface of cells, in engulfed endosomes, or in the cellular milieu. This allows cell to distinguish “self/healthy” and “non-self/unhealthy” [23-25]. After recognition and relevant ligand binding, PRRs can initiate nonspecific innate immune activities such as activating specific genes (notably IFNs), expression and secretion of specific sets of cytokines, initiating cell apoptosis or pyroptosis to induce inflammation and delay the spread of pathogens. The second important function of PPRs is activating adaptive immune response and modulating its activity [26].
Based on their protein domain homology, most PRRs in the innate immune system can fall into several categories: Toll-like receptors (TLRs), retinoic acid-inducible gene-I (RIG-I)-like receptors (RLRs), nucleotide oligomerization domain (NOD)-like receptors (NLRs), C-type lectin receptors (CLRs), absent in melanoma-2 (AIM2)-like receptors (ALRs) and cytosolic nucleic acid sensor cyclic GMP- AMP (cGAMP) synthase (cGAS) [27,28].
TLRs are highly conserved from the worm to mammals [16,29-33]. TLRs are type I integral membrane glycoproteins and consist of extracellular domains containing several leucine-rich-repeat motifs, which recognize PAMPs or DAMPs, the transmembrane domains, and a cytosolic signaling TIR domain that is responsible for downstream signal transduction [34-39]. There are 13 TLRs (TLR1-13), including 10 functional TLRs (TLR1-10) in human and 12 TLRs (TLR1-9, TLR11-13) expressed in mice [27]. TLRs have various expression patterns. Some TLRs (TLR1, 2, 4, 5, 6, 11) are expressed on the cell membranes in the form of heterodimers (TLR1/2, TLR2/6) or homodimers (TLR4, 5, 11), recognizing mainly bacterial lipids, lipoproteins, and lipopolysaccharides (LPS). Another set of TLRs (3, 7, 8, 9) are expressed in the subcellular organelles such as the endoplasmic reticulum (ER), endosomes, lysosomes, and endo-lysosomes, and recognize aberrant nucleic acids from viruses and other microorganisms. TLR3 recognizes double-stranded RNA viral metabolites in vesicles and on the cell surface, while TLR7 and 8 can recognize ssRNA inside of vesicles, chasing for viral infection. TLR9 is a vesicular sensor for DNA of viruses and a host DNA leakage [22]. Activated TLRs induce downstream IFN-I and NF-kB signaling pathways, resulting in the expression of proinflammatory cytokines, attraction to immune cells, and activation of type I and type III interferons signaling (see below) [40].
RLRs are the family of intracellular PRRs recognizing viral RNA in the cytoplasm [41]. The RLR family includes RIG-I, melanoma differentiation-associated gene 5 (MDA5), laboratory of genetics and physiology 2 (LGP2). In the N-terminus of RIG-1 and MDA5, two caspase activation and recruitment domains (CRAD) transmit signals to downstream interactors [42,43]. At the end of the C-terminus of RIG-I and LGP2, there is a repressor domain (RD) respectively [43,44], which inhibits the activation of the receptor [44,45]. Lacking RD, MDA5 does not have self-inhibitory function [43,46]. The middle DexD/H helicase domain functions as ATPase and helicase, and the C-terminal domain (CTD), which activates the protein itself with presence of viral RNA [47,48], are shared by all three RLRs. RIG-I can recognize dsRNA shorter than 1000 bp and 5’-triphosphate RNA of viruses, while MDA5 recognizes long-chain dsRNA that is greater than 1000 bp [49,50]. For SARS-CoV-2, RLRs should be a serious threat since both RIG-I and MDA5 are very effective against a few RNA and some DNA viruses (Table 1).
|
Baltimore classification |
Representative Virus |
RLR activated by the virus |
Reference |
|
I dsDNA |
Herpesviridae (herpes simplex virus type 1; Kaposi’s sarcoma-associated herpesvirus; Epstein–Barr virus) |
RIG-I, MDA5 |
[144-146] |
|
II dsDNA |
Not detected |
|
|
|
III dsRNA |
Reoviridae (rotavirus) |
RIG-I, MDA5 |
[147,148] |
|
IV ssRNA (+) |
Coronaviridae (SARS coronavirus) |
RIG-I, MDA5 |
[149-151] |
|
V ssRNA (–) |
Filoviridae (Ebola virus, Marburg virus) |
RIG-I, MDA5 |
[152,153] |
|
VI ssRNA (RT) |
Retroviridae (human immunodeficiency virus) |
RIG-I, MDA5 |
[154,155] |
|
VII dsDNA (RT) |
Hepadnaviridae (hepatitis B virus) |
RIG-I, MDA5 |
[156-158] |
NLRs are another group of intracellular PRRs responsible for bacterial identification [51-54]. Two signature receptors, NOD1 and NOD2, are critical for inflammatory signal transduction [55]. NOD1 recognizes the diaminopimelic acid (γ-D-glu-meso-diaminopimelic acid (iE-DAP)) in the cell wall of Gram-negative bacteria [56,57]. NOD2 can recognize muramyl dipeptide (MDP) in cell wall of all kinds of bacteria. Besides MDP, NOD2 can also recognize viral single-stranded RNA (ssRNA) [58,59]. Upon activation, NODs oligomerize and recruit the protein kinase RIP2 and CARD9 via CARD domains, leading to activation of NF-κB and MAP kinases. This induces secretion of IL-1b, IL-18, TNF, and other inflammatory signaling.
CLRs belong to phagocytic PRRs mainly expressed in monocytes, macrophages, Langerhans, and Dendritic cells (LCs and DCs). The receptors from CLR family bind carbohydrate moieties in a calcium-dependent manner using conserved carbohydrate recognition domains (CRDs) [60]. In contrast to other receptors, which induce signal transduction to activate the immune system, CLRs bind PAMPs and escort to cytoplasmic vesicles (lysosome or autophagosomes) for direct digestion and elimination, or to facilitate MHC-I presentation of the antigen [61-63].
ALRs include AIM2 and IFN-γ-inducible protein 16 (IFI16), which are discovered to recognize bacteria dsDNA through its C-terminal DNA-binding domain HIN-200 [64,65]. The N-terminal pyrin domain (PYD) interacts with the adaptor molecule, which is the apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (ASC), to activate both NF-κB and caspase-1 and induce inflammasomes formation and the maturation and secretion of IL-1β and IL-18 [64,66,67].
cGAS is a cytosolic double-stranded DNA (dsDNA) sensor that activates interferon responses through production of the second messenger cGAMP that activates the adaptor stimulator of interferon genes (STING), which is so called cGAS-cGAMP-STING pathway [28]. In human and other mammals, cGAS binds to the sugar-phosphate backbone of dsDNA coming from bacteria, DNA virus, retrovirus, dead cells, and even self-DNA, independently of the DNA sequences, which induces the conformation change of cGAS in its active site. Activated cGAS catalyzes 2'3'-cGAMP, which functions as a second messenger that binds to STING and leads to the translocation of STING from the ER to the ERGIC. STING is believed to induce the phosphorylation of TBK1, IKK and IRF3 after translocation, and therefore to induce interferon responses and signaling [28,68-70]. Recently, it was reported that cGAS like receptors (cGLRs) are nucleic acid receptors in Drosophila, which catalyze 3′2′-cGAMP and activate Sting-dependent antiviral responses [71,72].
Type I interferon pathway
Activation of any nucleic acid PRR sensors results in universal antiviral immune response, with an essential role of the type I and III interferon signaling. Type I interferon, or IFN-I, is the family of related genes among the first responders to viral infections. In humans, there are five major groups within the type I interferon family, designated IFN-α, β, κ, ε, and ω. Since virtually every cell in the body can produce IFNs, their expression is tightly controlled, and it is typically not secreted unless the cell becomes infected or encounters other types of stressors [22]. As discussed above, PRRs can be broadly divided into two groups. Sensors from the first group are expressed in the cytoplasm, consisting of nucleic acid sensors (NA sensors), which recognize immunogenic RNA (RIG-I, MDA5) or DNA molecules (cGAS, STING); and bacterial peptidoglycans sensors termed NOD1 and NOD2. The second group of sensors is the TLRs and CLRs. Activation of any of these receptors induces activation of key signaling kinase complexes TBK1/IKKε and IKK1/IKK2. While all membrane-bound receptors require adaptor proteins MyD88 or TRIF to activate downstream signaling kinases, cytosolic NA sensors (RIG-I, MDA5, cGAS, STING) activate TBK1/IKKε directly. Activated TBK1/IKKε complex phosphorylates members of IRF transcription factors family, while IKK1/IKK2 complex controls activation of NFκB transcription factors. While NFκB regulates the expression of inflammatory cytokines, IRF transcription factors are the key regulators of the type I IFN response leading to secretion of IFNs [73].
The type I IFN family molecules activate cells in both autocrine and paracrine manner, and in a self-promoting way. Secreted IFNs are recognized by the interferon-α receptor complex (IFNAR) consisting of two transmembrane proteins IFNAR1 and R2. Activation of IFNAR mediates downstream predominantly through the JAK/STAT signaling, which promotes further activation of type I IFN response, as well as inflammatory responses through NFκB pathway. Moreover, the type I IFN is closely linked to the cell apoptosis: under certain circumstances like viral infection, activation of the interferon-dependent pathways can promote apoptosis or pyroptosis of the cell. Molecules such as receptor-interacting protein kinase 1 (RIPK1), RIPK3, FAS-associated death domain protein (FADD), FLICE-like inhibitory protein (FLIP), and several caspases, which are essential regulators of different forms of cell death, are incorporated into signaling of TLRs, NOD-like receptor, and NA sensors [74]. These signaling modules have high capability to switch the cell status from inflammation to cell death, which is a core feature of IFN protection against viral infections.
Interferon provides effective protection from coronaviruses. Recent COVID-19 clinical data have shown that the IFN response in epithelial cells of the upper respiratory tract can curb SARS-CoV-2 replication. Some risk factors, for example, delayed or decreased interferon signaling [75], neutralizing of IFN-I by autoantibody [76], impropriate inflammatory cytokine responses [77] as well as the incidence of comorbidities (obesity, cardiovascular disease), higher expression of ACE2 in male and elderly [8], may favor developing more severe COVID-19 symptoms. Interestingly, such correlation is always shifted during early infections when the virus is still active [78]. Accordingly, SARS-CoV-2 viral proteins, directly or indirectly attempting to inhibit IFN-I response to gain extra time for safe replication. Trying to outsmart each other, host and virus show the typical pattern of the evolutional arms race when both partners engage multiple intelligent molecular tools in order to survive.
Host-Virus Arms Race
Like other viruses, SARS-CoV-2 produces a few PAMPs, including its ssRNA, dsRNA, S protein, and E protein, which can be directly recognized by the PPRs of innate immune system. Human cells can detect SARS-CoV-2 virus through several mechanisms (Figure 2). After injection or endocytosis, SARS-CoV-2’s genomic ssRNA can be recognized by endosomal TLR7 and TLR8 [8,79]; its intermediate dsRNA generated during viral replication is sensed by endosomal TLR3 and cytosolic RIG-I and MDA5 [80]; S protein may activate TLR4 [81,82], while E protein directly triggers TLR2 downstream signaling [83]. Upon PAMPs recognition, membranous PRRs activate downstream signaling through specific adaptor proteins are utilized by (MyD88 for all TLRs, TRIF for TLR3; IRIF, TRAM for TIR4) to active NF-κB, MAPK, and IFN-I pathways. Correspondently, activated transcription factors (RelA, IRS3/7, AP-1) induces expression and secretion of proinflammatory cytokines and chemokines (such as IL-6, TNF pro–IL-1β and IL-8), type I (mostly α and β) and type III (γ1/2/3) IFNs [40,84]. In the cytoplasm, the presence of viral RNA metabolites could be recognized by RIG-I and MDA5, which activates IFN-I response through signaling kinases IKKε/TBK1 [80].
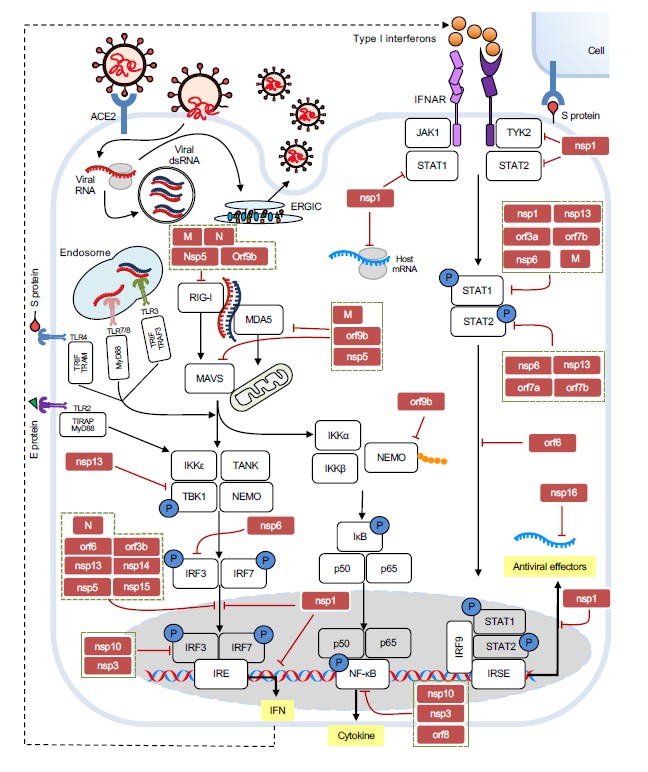
Figure 2. Replication cycle of SARS-CoV-2 and the interplay between host innate immunity and the virus. SARS-CoV-2 enters the cell via ACE2. Viral RNA gets translated by host ribosomes and replicated within the double-membrane vesicles. The virions are assembled at ERGIC and then egressed outside the cell. Host cells detect viral proteins and nucleic acids through PRRs and induce interferon reaction and other antiviral responses through relevant pathways. White squares represent each protein participating in the antiviral responses and the blue circles indicate phosphorylation. Red squares and red bar-headed lines stand for the viral proteins that inhibit the related innate immune response.
Besides PAMPs recognition, direct dsRNA-induced activation through RNA-activated protein kinase (PKR) and 2’-5’-oligoadenylate synthetase-like (OASL) was observed in infected SARS-CoV-2 respiratory epithelial derived cells and cardiomyocytes [85]. In addition to the rapid interferon and inflammatory response against the PAMPs, the viral infection could lead to host cell apoptosis followed by releasing self-DNA from the nucleus into the cytoplasm, which is one of DAMPs and can directly trigger cGAS/STING and AIM2/IFI16 pathways [80]. The innate immune system recognizes pathogens and activates the adaptive immune response, including activation of T cells and B cells [26]. Therefore, the virus has very little space to succeed if innate immunity is fully active.
However, the SARS-CoV-2 virus can directly inhibit multiple pathways of innate immune responses at all stages of infection. In addition to their essential roles in the viral life cycle, most SARS-CoV-2 viral proteins antagonize core cellular functions in human cells to evade host immune responses in the favor of the virus (comprehensive summary, see Figure 2 and Table 2).
|
SARS-CoV-2 proteins |
Target |
Mechanism |
References |
|
Nsp1 |
40S ribosomal and primosomal subunit; 18S rRNA; STAT1; IRF3; STAT2; Tyk2 |
Translation inhibition by interfering with host mRNA binding and nuclear export; blocks IRF3 nuclear translocation; inhibition of STAT1 phosphorylation; reduced expression of STAT2 and Tyk2 |
[86-88,91-98] |
|
Nsp3 |
ISG15, IRF3, PARP9/DTX3L, NF-kB |
Cleaves ISG15 from IRF3; inhibits IFN-I promoters, IRF3 and NF-kB binding sites; reverses PARP9/DTX3L-dependent ADP-ribosylation |
[99-101] |
|
Nsp5 |
RIG-I; MAVS; IRF3; STAT1; STAT2 |
Cleaves off the 10 most-N-terminal amino acids from RIG-I; promotes the ubiquitination and proteosome-mediated degradation of MAVS; inhibits blocking the nucleus translocation of phosphorylated IRF3; induces phospho-STAT1/2 accumulation impairing type I IFN signaling |
[100,102-104] |
|
Nsp6 |
IRF3, TBK1; STAT1; STAT2 |
Suppresses phosphorylation of IRF3 by binding to TBK1; inhibits STAT1 and STAT2 phosphorylation |
[95] |
|
Nsp8 |
7SL RNA Component of SRP54 |
disrupts protein trafficking for secretion or membrane integration |
[88] |
|
Nsp9 |
7SL RNA Component of SRP19 |
disrupts protein trafficking for secretion or membrane integration |
[88] |
|
Nsp10 |
IRF3; NF-kB |
Impairs the activity of IFNA4 and IFNB1, IRF3 binding and NF-kB binding, and suppresses cytokines production |
[100] |
|
Nsp13 |
TBK1; STAT1; STAT2; IRF3 |
Inhibits TBK-1, STAT1 and STAT2 phosphorylation; blocks IRF3 nuclear translocation |
[95,100,109] |
|
Nsp14 |
IRF3; IFNAR1 |
Inhibits IRF3 nuclear translocation; induces lysosomal degradation of IFNAR1; inhibits host cellular translation via ExoN and N7-MTase activities |
[100,109,111] |
|
Nsp15 |
IRF3; early autophagosome |
Inhibits IRF3 nuclear translocation; inhibits de novo autophagy induction |
[100] |
|
Nsp16 |
U1 and U2 splicing RNAs |
Suppresses host mRNA splicing through binding to the pre-mRNA recognition domains of the U1 and U2 splicing RNAs |
[88] |
|
S |
ACE2 |
Evades host cells and induces syncytia formation through binding to ACE2 receptor |
[126-128] |
|
E |
Autophagosome |
Blocks autophagic turnover |
[100] |
|
M |
MAVS; autophagosome; RIG-I/MDA-5 |
Impairs MAVS aggregation and recruitment of downstream components; induces LC3B accumulation in the perinuclear space; suppresses type I and III IFN expression by targeting RIG-I/MDA-5 signaling |
[100,112,113] |
|
N |
RIG-I; TBK1/IRF3 |
Binds to with the RIG-I protein through its DExD/H domain and suppresses IFN-β production; impairs TBK1/IRF3 association and IRF3 nuclear translocation |
[114,115] |
|
Orf3a |
STAT1; lysosomes with autophagosomes |
Inhibits STAT1 phosphorylation, blocking the fusion of lysosomes with autophagosomes |
[95,100] |
|
Orf3b |
IRF3 |
Inhibits IRF3 nuclear translocation |
[116] |
|
Orf6 |
KPNA2; STAT1; IRF3; ISGF3 |
Interacts with KPNA2; blocks STAT1, IRF3 and ISGF3 nuclear translocation |
[95,109,117,118] |
|
Orf7a |
STAT2; lysosomes |
Inhibits STAT2 phosphorylation; decreases lysosomes acidification |
[95,100] |
|
Orf7b |
STAT1; STAT2 |
Inhibits STAT1 and STAT2 phosphorylation |
[95] |
|
Orf8 |
MHC-Ι molecules; NF-κB |
Mediates their lysosomal degradation of MHC-I molecules; inhibits NF-κB-responsive promoter |
[109,119,123] |
|
Orf9b |
NEMO; RIG-I; MDA-5; MAVS; TRIF; STING; TBK1; IRF3 |
Interrupts K63-linked polyubiquitination of NEMO and inhibits IFN signaling; interacts with RIG-I, MDA-5, MAVS, TRIF, STING, and TBK1 and impedes the phosphorylation and nuclear translocation of IRF3 |
[120,121] |
For example, from the molecular level of host antiviral response, right after it is released from the pp1a and pp1ab, nsp1 binds to 18S ribosomal RNA and 40S subunit at the mRNA entry channel of the ribosome, which results in global inhibition of mRNA translation upon viral infection, to earn extra time for the virus to replicate itself before the cell can shut down translation machinery or induce self-degradation [86-92]. Not only that, nsp1 blocks nuclear export of host mRNAs, including IFN, by preventing the proper binding of NXF1 to mRNA export adaptors and NXF1 docking at the nuclear pore complex [93,94]. Nsp1 is also able to inhibit IFN signaling in part by blocking STAT1, IRF3 phosphorylation, Tyk2, and STAT2 activation [95,96], and hypothetically –through enigmatic interaction with the primosomal proteins POLA1, POLA2, PRIM1, PRIM2 [87,97]. It was reported that the 500-532 deletion in the nsp1 coding region leads to lower IFN-I response, which suggests nsp1 is closely associated with immune counteraction [98]. Nsp3, the papain-like protease, preferentially cleaves the ubiquitin-like interferon-stimulated gene 15 protein (ISG15) from interferon responsive factor 3 (IRF3), thereby inhibits type I interferon responses [99]. It’s also reported that nsp3 can strongly impair the activity of IFNA4 and IFNB1, IRF3 binding and NF-κB binding [100]. Nsp3’s macrodomain can also reverse the downstream PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling [101]. Nsp5 inhibits dsRNA induced IFN induction from RIG-I–mitochondrial antiviral signaling (MAVS) protein–IFN pathway by cleaving off the 10 most-N-terminal amino acids from RIG-I and suppress its ability to activate MAVS, also promoting the ubiquitination and proteosome-mediated degradation of MAVS [102]. Furthermore, nsp5 was reported to suppress INF-β transcription induced by TBK1 and IKKε via blocking the nuclear translocation of phosphorylated IRF3 and impairing type I IFN signaling by inducing phospho-STAT1/2 accumulation [100,103,104]. Nsp6 suppresses phosphorylation of IRF3 by binding and inhibiting TANK binding kinase 1 (TBK1) phosphorylation; it also impairs phosphorylation of STAT1 and STAT2 (95). Nsp8 and nsp9 interact with 7SL RNA component of SRP54 and SRP19, the components of the signal recognition particle (SRP), which recognizes the signal peptide of secretory proteins. By disrupting protein trafficking, these two nsps affect cytokine secretion and MHC-I recycling and induce significant reduction of the immune response [88].
Another nonstructural protein, nsp10, was reported to impair the activity of type I interferons by interacting with IRF3 and NF-κB signaling and suppressing cytokines production upon infection [100]. This activity is probably mediated by the nsp10’s RNA 2-0-methylation activity, known as immunosuppressive RNA modification, that allows the virus to avoid RIG-I/MDA-5 recognition [105,106]. Nsp12 was reported attenuating type I interferon production by inhibiting IRF3 nuclear translocation [107], whereas other data suggest otherwise [108]. The nsp13 protein downregulates primary interferon production by limiting the nuclear localization of IRF3. Also, nsp13 can interact with signaling kinase TBK1, and block its activation and phosphorylation of transcription factors STAT1 and STAT2 [87,95,100,109]. Furthermore, nsp13 is involved in viral RNA 5’ cap synthesis – a modification that must be present on the 5’ end of every mRNA to avoid recognition by RIG-I [110].
The nsp14 protein inhibits activation in a different manner –through activation of lysosomal degradation of the essential interferon receptor IFNAR1. Lower number of IFN receptors slows down autocrine and paracrine activation of STATs transcription factors and results to weaker immunity [100,109]. Nsp14 in complex with nsp10 can also distort host translation processes due to their exonuclease and N7-MTase activities [111]. SARS-CoV-2 nsp15 was documented to suppress primary interferon production and interferon signaling by inhibiting the nuclear localization of IRF3. Nsp16 binds to the pre-mRNA recognition domains of the U1 and U2 splicing RNAs and suppresses host mRNA splicing during SARS-CoV-2 infection [88]. It is also believed that nsp16 modifies with methyl group the cap of viral RNA for evading RIG-I detection [87].
Not only non-structural proteins can play against PRR sensors. The structural M protein interacts with the central adaptor protein MAVS in the innate immune response pathways, impairing MAVS aggregation and its recruitment of downstream TRAF3, TBK1, as well as the IRF3 transcription factor, resulting in impaired antiviral response [112]. Another study reported that SARS-CoV-2’s M protein also suppresses type I and III IFN expression by targeting RIG-I/MDA-5 signaling to attenuate antiviral immunity and enhance viral replication [113]. The N protein of SARS-CoV-2 inhibits TRIM25/RIG-I interaction through binding to the RIG-I protein through its DExD/H domain. And N protein also impairs TBK1/IRF3 association, therefore prevents nuclear translocation of IRF3 and IFN expression [114,115]. Accessory protein orf3b functions as a potent IFN antagonist, of which the suppression of IFN induction depends on the length its C terminus, which prevents the nuclear translocation of IRF3 [116]. Orf6 localizes at the nuclear pore complex and directly interacts with Nup98-Rae1 through its C-terminal domain to impair the docking process of cargo-receptor (karyopherin/importin) complex, thereby blocks STAT1, IRF3 and ISGF3 nuclear translocation. Whereas it was reported that orf6 inhibits IRF3 nuclear translocation via interacting with importin Karyopherin a2 (KPNA2) [95,109,117,118]. Another accessory protein, orf7a, suppresses the IFN-I response by inhibiting STAT2 phosphorylation, while orf7b inhibits the phosphorylation of both STAT1 and STAT2 [95]. Orf8 can strongly inhibit type I interferon (IFN-β) and NF-κB-responsive promoter, as well as the interferon-stimulated response element (ISRE) [109,119]. Orf9b accumulates immediately after released during SARS-CoV-2 infection and inhibits the RIG-I/MAVS pathway dependent type I interferon response by interrupting the K63-linked polyubiquitination of the interferon signaling modulator NEMO [120]. Orf9b also was reported to interact with RIG-I, MDA-5, MAVS, TRIF, STING, and TBK1 and impede the phosphorylation and nuclear translocation of IRF3 [121].
Besides direct molecular confrontations, SARS-CoV-2 also affects macromolecular processes, modulating cell autophagy, fusion, and cell death program. Notably, after infection, virus generates double-membrane vesicles (DMVs) as its viral replication organelles to physically separate viral biochemistry from cellular cytoplasm, in this way avoiding PRRs recognition [122].
Autophagy plays an important role in regulating immunity-related cell death and antiviral responses. Similar to blunting PRR signaling, SARS-CoV-2 is attempting to distort autophagy as well. The nsp15 protein inhibits de novo autophagy induction [100], while the E and M structural proteins were reported to block autophagic turnover in the host cells to prevent degradation of assembled virions [100]. Autophagy is also modulated by SARS-CoV-2 accessory proteins, like orf3a, which inhibits autophagic turnover by targeting the late endosomes and blocking the fusion with autophagosomes [100]. Another protein, orf7a, blocks autophagic turnover by affecting lysosomal acidification [100]. The orf8 protein directly interacts with MHC-Ι molecules and mediates their lysosomal degradation via autophagy. Therefore, SARS-CoV-2–infected cells are much less sensitive to be lysed by cytotoxic T lymphocytes [123].
Finally, SARS-CoV-2 could induce cells fusion – another mechanism of viral spreading. During the virus assembly process, S protein with other structural proteins (E, M, N) are translocated into the lumen of the intermediate compartment at the endoplasmic reticulum (ER)-Golgi interface [124]. Then, the matured virions traffic to de-acidified lysosomes and egress by Arl8b-dependent lysosomal exocytosis to start another round of infection [125]. However, S protein also translocates to the surface of infected cells through the COPI (retrograde) and COPII (anterograde) transport, leading to increased TMEM16F expression, which in turn increases the phosphatidylserine concentration on the plasma membrane. The interaction between S protein on the infected cell surface with the ACE2 receptor on the neighboring cell, as well as the increased concentration of phosphatidylserine, may induce syncytia formation. In this manner, SARS-CoV-2 could spread through the tissue remaining inside the cells, out of reach for adaptive immunity [126-128]. Notably, in terms of the arms race, the evolution of the S protein contributes to the escape from the adaptive immune system, helping the virus to extend replication time [129]. Indeed, other than the S protein, it was reported that the Alpha (B.1.1.7) variant has dramatically increased protein levels of N, orf9b and orf6 and sgRNAs, which makes it more effectively dampen down epithelial cell innate immune responses in the airway [130].
Beyond the Nature: Antiviral Pharmaceuticals as a Novel Arm to Slow Down the Virus
Such complicated viral counterintelligence aims to blunt host’s innate immunity and earn extra time for its replication. However, humans have one more option – pharmaceuticals. The viral protease and RNA-dependent RNA polymerase have been proven to be the bottleneck of many viruses, and pharmaceutic drugs could enhance PRRs sensing by inducing chimeric viral metabolites accumulation in the infected cells. Indeed, uncut polyprotein or not finished genomic RNA polymers could be easy baits for PRRs [11].
Viruses generate a variety of aberrant nucleic acids during their distinctive replication cycles, which can be recognized by relevant PRRs leading to activation of type I/III interferon signaling. Therefore, it is not surprise that the most effective anti-COVID clinical drugs target viral replication and translation processes (Figure 3, Table 3). Any delay in replication makes the virus vulnerable to immune clearance when its double-stranded RNA complexes are exposed to cytosolic PRRs. Therefore, there is no surprise that even drugs with weak pharmaceutical activity toward SARS-CoV-2, such as Lopinavir/Ritonavir (HIV 3CLpro inhibitors), Remdesivir (originally developed for the treatment of Ebola virus) [131,132], Oseltamivir and Enisamium iodide (influenza drugs, preventing severe development of COVID-19) [132-136], similar to specific inhibitors like Molnupiravir [137-140] and Paxlovid [141] work the best at the early stages of infections, when multiplication of viruses occurs. Multiple clinical trials showed that inhibitors of RdRp and viral proteases, taken during the first 3-4 days of infection, dramatically improved the chances of patients to avoid severe or long COVID-19. Given that these inhibitors impair RdRp or protease activity, they can increase the concentration of abnormal viral RNA and proteins to the levels visible for inhibited PRR/IFN-I pathways. This could facilitate immune clearance and even pre-activate cells before invasion. For example, Molnupiravir in several preclinical models has been shown to be active not only for the treatment, but also for prophylaxis and prevention not only SARS-CoV-2, but also SARS-CoV and MERS [137,142].
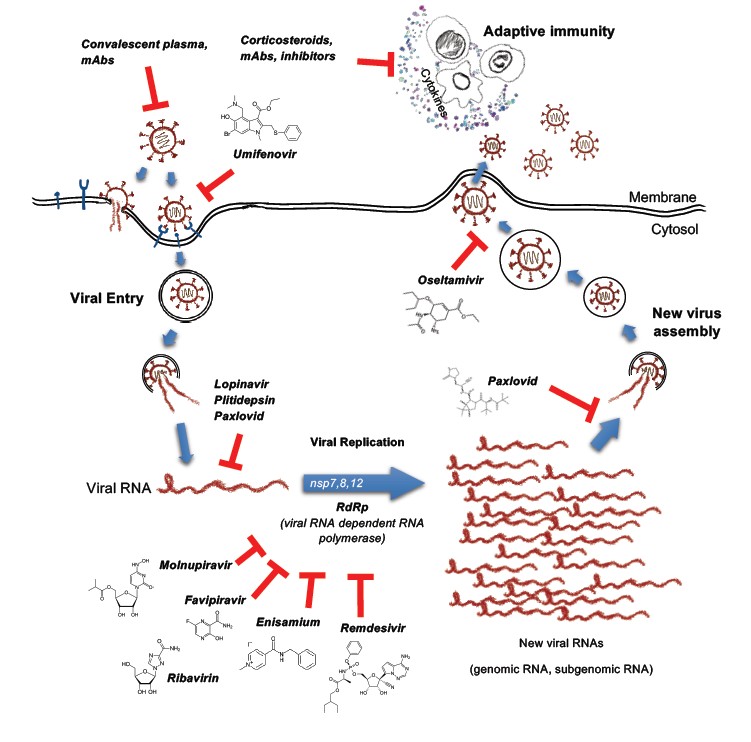
|
Drug |
Target |
|
Molnupiravir |
RdRp inhibitor |
|
Paxlovid |
Protease inhibitor |
|
Remdesivir |
RdRp inhibitor |
|
Favipiravir |
RdRp inhibitor |
|
Oseltamivir |
RdRp inhibitor |
|
Enisamium iodide |
RdRp inhibitor |
|
Plitidepsin |
Transcription inhibitor |
|
Lopinavir/Ritonavir |
Protease inhibitor |
Altogether, antiviral drugs seem to help PRR in the detection of the infectious agent, while the well-timed interferon response appears to be the key factor preventing severe development of the COVID-19. However, the arms race is underway – once a new drug is released, the virus will rapidly start another round of evolution. Therefore, innate immunity and its multiple antiviral sensors remain humans' main ally in the pandemic world.
Conclusions and Future Outlook
It has been solidly confirmed by numerous studies that timely innate immunity responses play a critical role in fighting SARS-CoV-2 infection. For example, a fundamental difference between adults and children helps to explain why kids are less likely to become seriously ill from SARS-CoV-2. According to recent study [143], the more robust innate immune response in the airways through activation of PRR-Interferon signaling helped restrict viral replication early. Older or susceptible people often possess less active innate immunity, and lack of timely interferon response is associated with higher viral load, longer course of infection, and more possibilities for various organs invasion as well as multi-organs failure [143]. Therefore, to understand the complicated interplay between the host immunity and SARS-CoV-2 is paramount to identifying the best-suited approaches for individual patients at a given time.
This review provides a systematic overview on the biological foundations of the life cycle of SARS-CoV-2 and the immune arms race between the host and the virus. PRR recognitions and subsequential interferon responses and signaling is the first line to control the viral infection. We also discuss a few promising antiviral pharmaceuticals based on their targets, protease inhibition or RdRp blockade. To apply those drugs at the early stages of COVID-19 is the most workable strategy to control the disease.
Nevertheless, compared to the dramatically increasing number of the patients, there are still not many pharmaceuticals available for COVID-19 treatment, less if being financially affordable is considered. Future study efforts should lie in more detailed investigations of the molecular biology of SARS-CoV-2 and discovery of both novel and repurposed drugs.
Funding
This work is supported by the Science and Technology project affiliated to the education department of Chongqing municipality (KJQN202100436).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to this work, and approved it for publication.
References
2. Payne S. Family Coronaviridae. Viruses2017. p. 149-58.
3. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367(6485):1444-8.
4. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5(4):562-9.
5. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-92 e6.
6. Cantuti-Castelvetri L, Ojha R, Pedro LD, Djannatian M, Franz J, Kuivanen S, et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science. 2020;370(6518):856-60.
7. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181(2):271-80 e8.
8. Christie MJ, Irving AT, Forster SC, Marsland BJ, Hansbro PM, Hertzog PJ, et al. Of bats and men: Immunomodulatory treatment options for COVID-19 guided by the immunopathology of SARS-CoV-2 infection. Sci Immunol. 2021;6(63):eabd0205.
9. Peacock TP, Goldhill DH, Zhou J, Baillon L, Frise R, Swann OC, et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat Microbiol. 2021;6(7):899-909.
10. Romano M, Ruggiero A, Squeglia F, Maga G, Berisio R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells. 2020;9(5).
11. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779-82.
12. Angelini MM, Akhlaghpour M, Neuman BW, Buchmeier MJ. Severe acute respiratory syndrome coronavirus nonstructural proteins 3, 4, and 6 induce double-membrane vesicles. mBio. 2013;4(4).
13. Snijder EJ, Limpens R, de Wilde AH, de Jong AWM, Zevenhoven-Dobbe JC, Maier HJ, et al. A unifying structural and functional model of the coronavirus replication organelle: Tracking down RNA synthesis. PLoS Biol. 2020;18(6):e3000715.
14. Klein S, Cortese M, Winter SL, Wachsmuth-Melm M, Neufeldt CJ, Cerikan B, et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun. 2020;11(1):5885.
15. Janeway CA, Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol. 1989;54 Pt 1:1-13.
16. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783-801.
17. Brubaker SW, Bonham KS, Zanoni I, Kagan JC. Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol. 2015;33:257-90.
18. Herwald H, Egesten A. On PAMPs and DAMPs. J Innate Immun. 2016;8(5):427-8.
19. Zindel J, Kubes P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu Rev Pathol. 2020;15:493-518.
20. Pandolfi F, Altamura S, Frosali S, Conti P. Key Role of DAMP in Inflammation, Cancer, and Tissue Repair. Clin Ther. 2016;38(5):1017-28.
21. Gasteiger G, D'Osualdo A, Schubert DA, Weber A, Bruscia EM, Hartl D. Cellular Innate Immunity: An Old Game with New Players. Journal of Innate Immunity. 2017;9(2):111-25.
22. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373-84.
23. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14(10):1014-22.
24. Cen X, Liu S, Cheng K. The Role of Toll-Like Receptor in Inflammation and Tumor Immunity. Front Pharmacol. 2018;9:878.
25. Amarante-Mendes GP, Adjemian S, Branco LM, Zanetti LC, Weinlich R, Bortoluci KR. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front Immunol. 2018;9:2379.
26. Sonnenberg GF, Hepworth MR. Functional interactions between innate lymphoid cells and adaptive immunity. Nat Rev Immunol. 2019;19(10):599-613.
27. Li D, Wu M. Pattern recognition receptors in health and diseases. Signal Transduct Target Ther. 2021;6(1):291.
28. Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786-91.
29. Janeway CA, Jr., Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197-216.
30. Hoffmann JA. The immune response of Drosophila. Nature. 2003;426(6962):33-8.
31. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499-511.
32. Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004;430(6996):257-63.
33. Beutler B. Innate immunity: an overview. Mol Immunol. 2004;40(12):845-59.
34. Bowie A, O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: signal generators for pro-inflammatory interleukins and microbial products. J Leukoc Biol. 2000;67(4):508-14.
35. Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. A46R and A52R from vaccinia virus are antagonists of host IL-1 and toll-like receptor signaling. Proc Natl Acad Sci U S A. 2000;97(18):10162-7.
36. Enkhbayar P, Kamiya M, Osaki M, Matsumoto T, Matsushima N. Structural principles of leucine-rich repeat (LRR) proteins. Proteins. 2004;54(3):394-403.
37. Kim HM, Park BS, Kim JI, Kim SE, Lee J, Oh SC, et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell. 2007;130(5):906-17.
38. Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, et al. Crystal structure of the TLR1-TLR2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell. 2007;130(6):1071-82.
39. Gao D, Li W. Structures and recognition modes of toll-like receptors. Proteins. 2017;85(1):3-9.
40. Carty M, Guy C, Bowie AG. Detection of Viral Infections by Innate Immunity. Biochem Pharmacol. 2021;183:114316.
41. Onomoto K, Onoguchi K, Yoneyama M. Regulation of RIG-I-like receptor-mediated signaling: interaction between host and viral factors. Cell Mol Immunol. 2021;18(3):539-55.
42. Gee P, Chua PK, Gevorkyan J, Klumpp K, Najera I, Swinney DC, et al. Essential Role of the N-terminal Domain in the Regulation of RIG-I ATPase Activity. Journal of Biological Chemistry. 2008;283(14):9488-96.
43. Jiang F, Ramanathan A, Miller MT, Tang GQ, Gale M, Jr., Patel SS, et al. Structural basis of RNA recognition and activation by innate immune receptor RIG-I. Nature. 2011;479(7373):423-7.
44. Xu XX, Wan H, Nie L, Shao T, Xiang LX, Shao JZ. RIG-I: a multifunctional protein beyond a pattern recognition receptor. Protein Cell. 2018;9(3):246-53.
45. Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. Structural insights into RNA recognition by RIG-I. Cell. 2011;147(2):409-22.
46. Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99(2):637-42.
47. Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, et al. The C-terminal regulatory domain is the RNA 5'-triphosphate sensor of RIG-I. Mol Cell. 2008;29(2):169-79.
48. Kowalinski E, Lunardi T, McCarthy AA, Louber J, Brunel J, Grigorov B, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147(2):423-35.
49. Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601-10.
50. Wang Y, Ludwig J, Schuberth C, Goldeck M, Schlee M, Li H, et al. Structural and functional insights into 5'-ppp RNA pattern recognition by the innate immune receptor RIG-I. Nat Struct Mol Biol. 2010;17(7):781-7.
51. Inohara N, Nunez G. NODs: intracellular proteins involved in inflammation and apoptosis. Nat Rev Immunol. 2003;3(5):371-82.
52. Fritz JH, Ferrero RL, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat Immunol. 2006;7(12):1250-7.
53. Meunier E, Broz P. Evolutionary Convergence and Divergence in NLR Function and Structure. Trends Immunol. 2017;38(10):744-57.
54. Alvarez CA, Ramirez-Cepeda F, Santana P, Torres E, Cortes J, Guzman F, et al. Insights into the diversity of NOD-like receptors: Identification and expression analysis of NLRC3, NLRC5 and NLRX1 in rainbow trout. Mol Immunol. 2017;87:102-13.
55. Lu Y, Zheng Y, Coyaud E, Zhang C, Selvabaskaran A, Yu Y, et al. Palmitoylation of NOD1 and NOD2 is required for bacterial sensing. Science. 2019;366(6464):460-7.
56. Chamaillard M, Hashimoto M, Horie Y, Masumoto J, Qiu S, Saab L, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol. 2003;4(7):702-7.
57. Pashenkov MV, Dagil YA, Pinegin BV. NOD1 and NOD2: Molecular targets in prevention and treatment of infectious diseases. Int Immunopharmacol. 2018;54:385-400.
58. Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem. 2003;278(11):8869-72.
59. Sabbah A, Chang TH, Harnack R, Frohlich V, Tominaga K, Dube PH, et al. Activation of innate immune antiviral responses by Nod2. Nat Immunol. 2009;10(10):1073-80.
60. Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465-79.
61. Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol Rev. 2014;262(1):193-215.
62. Dambuza IM, Brown GD. C-type lectins in immunity: recent developments. Curr Opin Immunol. 2015;32:21-7.
63. Murugaiah V, Tsolaki AG, Kishore U. Collectins: Innate Immune Pattern Recognition Molecules. Adv Exp Med Biol. 2020;1204:75-127.
64. Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. 2009;458(7237):514-8.
65. Feng S, Chen T, Lei G, Hou F, Jiang J, Huang Q, et al. Absent in melanoma 2 inflammasome is required for host defence against Streptococcus pneumoniae infection. Innate Immun. 2019;25(7):412-9.
66. Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB. The AIM2-like Receptors Are Dispensable for the Interferon Response to Intracellular DNA. Immunity. 2016;45(2):255-66.
67. Wang B, Tian Y, Yin Q. AIM2 Inflammasome Assembly and Signaling. Adv Exp Med Biol. 2019;1172:143-55.
68. Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell. 2013;51(2):226-35.
69. Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell. 2014;54(2):289-96.
70. Chen Q, Sun L, Chen ZJ. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol. 2016;17(10):1142-9.
71. Slavik KM, Morehouse BR, Ragucci AE, Zhou W, Ai X, Chen Y, et al. cGAS-like receptors sense RNA and control 3'2'-cGAMP signalling in Drosophila. Nature. 2021;597(7874):109-13.
72. Holleufer A, Winther KG, Gad HH, Ai X, Chen Y, Li L, et al. Two cGAS-like receptors induce antiviral immunity in Drosophila. Nature. 2021;597(7874):114-8.
73. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36-49.
74. Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol. 2014;14(9):601-18.
75. Channappanavar R, Fehr AR, Vijay R, Mack M, Zhao J, Meyerholz DK, et al. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe. 2016;19(2):181-93.
76. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science. 2020;370(6515).
77. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol. 2020;5(49).
78. Cheemarla NR, Watkins TA, Mihaylova VT, Wang B, Zhao D, Wang G, et al. Dynamic innate immune response determines susceptibility to SARS-CoV-2 infection and early replication kinetics. J Exp Med. 2021;218(8).
79. van der Made CI, Simons A, Schuurs-Hoeijmakers J, van den Heuvel G, Mantere T, Kersten S, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA. 2020;324(7):663-73.
80. Mdkhana B, Saheb Sharif-Askari N, Ramakrishnan RK, Goel S, Hamid Q, Halwani R. Nucleic Acid-Sensing Pathways During SARS-CoV-2 Infection: Expectations versus Reality. J Inflamm Res. 2021;14:199-216.
81. Choudhury A, Mukherjee S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J Med Virol. 2020;92(10):2105-13.
82. Shirato K, Kizaki T. SARS-CoV-2 spike protein S1 subunit induces pro-inflammatory responses via toll-like receptor 4 signaling in murine and human macrophages. Heliyon. 2021;7(2):e06187.
83. Zheng M, Karki R, Williams EP, Yang D, Fitzpatrick E, Vogel P, et al. TLR2 senses the SARS-CoV-2 envelope protein to produce inflammatory cytokines. Nat Immunol. 2021;22(7):829-38.
84. Amor S, Fernandez Blanco L, Baker D. Innate immunity during SARS-CoV-2: evasion strategies and activation trigger hypoxia and vascular damage. Clin Exp Immunol. 2020;202(2):193-209.
85. Li Y, Renner DM, Comar CE, Whelan JN, Reyes HM, Cardenas-Diaz FL, et al. SARS-CoV-2 induces double-stranded RNA-mediated innate immune responses in respiratory epithelial-derived cells and cardiomyocytes. Proc Natl Acad Sci U S A. 2021;118(16).
86. Thoms M, Buschauer R, Ameismeier M, Koepke L, Denk T, Hirschenberger M, et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369(6508):1249-55.
87. Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583(7816):459-68.
88. Banerjee AK, Blanco MR, Bruce EA, Honson DD, Chen LM, Chow A, et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell. 2020;183(5):1325-39 e21.
89. Schubert K, Karousis ED, Jomaa A, Scaiola A, Echeverria B, Gurzeler LA, et al. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat Struct Mol Biol. 2020;27(10):959-66.
90. Kilkenny ML, Veale CE, Guppy A, Hardwick SW, Chirgadze DY, Rzechorzek NJ, et al. Structural basis for the interaction of SARS-CoV-2 virulence factor nsp1 with DNA polymerase alpha-primase. Protein Sci. 2021.
91. Mendez AS, Ly M, Gonzalez-Sanchez AM, Hartenian E, Ingolia NT, Cate JH, et al. The N-terminal domain of SARS-CoV-2 nsp1 plays key roles in suppression of cellular gene expression and preservation of viral gene expression. Cell Rep. 2021;37(3):109841.
92. Yuan S, Peng L, Park JJ, Hu Y, Devarkar SC, Dong MB, et al. Nonstructural Protein 1 of SARS-CoV-2 Is a Potent Pathogenicity Factor Redirecting Host Protein Synthesis Machinery toward Viral RNA. Mol Cell. 2020;80(6):1055-66 e6.
93. Burke JM, St Clair LA, Perera R, Parker R. SARS-CoV-2 infection triggers widespread host mRNA decay leading to an mRNA export block. RNA. 2021;27(11):1318-29.
94. Zhang K, Miorin L, Makio T, Dehghan I, Gao S, Xie Y, et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci Adv. 2021;7(6).
95. Xia H, Cao Z, Xie X, Zhang X, Chen JY, Wang H, et al. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020;33(1):108234.
96. Kumar A, Ishida R, Strilets T, Cole J, Lopez-Orozco J, Fayad N, et al. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J Virol. 2021;95(13):e0026621.
97. Starokadomskyy P, Escala Perez-Reyes A, Burstein E. Immune Dysfunction in Mendelian Disorders of POLA1 Deficiency. J Clin Immunol. 2021;41(2):285-93.
98. Lin JW, Tang C, Wei HC, Du B, Chen C, Wang M, et al. Genomic monitoring of SARS-CoV-2 uncovers an Nsp1 deletion variant that modulates type I interferon response. Cell Host Microbe. 2021;29(3):489-502 e8.
99. Shin D, Mukherjee R, Grewe D, Bojkova D, Baek K, Bhattacharya A, et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587(7835):657-62.
100. Hayn M, Hirschenberger M, Koepke L, Nchioua R, Straub JH, Klute S, et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021;35(7):109126.
101. Russo LC, Tomasin R, Matos IA, Manucci AC, Sowa ST, Dale K, et al. The SARS-CoV-2 Nsp3 macrodomain reverses PARP9/DTX3L-dependent ADP-ribosylation induced by interferon signaling. J Biol Chem. 2021;297(3):101041.
102. Liu Y, Qin C, Rao Y, Ngo C, Feng JJ, Zhao J, et al. SARS-CoV-2 Nsp5 Demonstrates Two Distinct Mechanisms Targeting RIG-I and MAVS To Evade the Innate Immune Response. mBio. 2021;12(5):e0233521.
103. Wu Y, Ma L, Zhuang Z, Cai S, Zhao Z, Zhou L, et al. Main protease of SARS-CoV-2 serves as a bifunctional molecule in restricting type I interferon antiviral signaling. Signal Transduct Target Ther. 2020;5(1):221.
104. Fung SY, Siu KL, Lin H, Yeung ML, Jin DY. SARS-CoV-2 main protease suppresses type I interferon production by preventing nuclear translocation of phosphorylated IRF3. Int J Biol Sci. 2021;17(6):1547-54.
105. Daffis S, Szretter KJ, Schriewer J, Li J, Youn S, Errett J, et al. 2'-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468(7322):452-6.
106. Hyde JL, Diamond MS. Innate immune restriction and antagonism of viral RNA lacking 2-O methylation. Virology. 2015;479-480:66-74.
107. Wang W, Zhou Z, Xiao X, Tian Z, Dong X, Wang C, et al. SARS-CoV-2 nsp12 attenuates type I interferon production by inhibiting IRF3 nuclear translocation. Cell Mol Immunol. 2021;18(4):945-53.
108. Li A, Zhao K, Zhang B, Hua R, Fang Y, Jiang W, et al. SARS-CoV-2 NSP12 Protein Is Not an Interferon-beta Antagonist. J Virol. 2021;95(17):e0074721.
109. Yuen CK, Lam JY, Wong WM, Mak LF, Wang X, Chu H, et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg Microbes Infect. 2020;9(1):1418-28.
110. Devarkar SC, Wang C, Miller MT, Ramanathan A, Jiang F, Khan AG, et al. Structural basis for m7G recognition and 2'-O-methyl discrimination in capped RNAs by the innate immune receptor RIG-I. Proc Natl Acad Sci U S A. 2016;113(3):596-601.
111. Hsu JC, Laurent-Rolle M, Pawlak JB, Wilen CB, Cresswell P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc Natl Acad Sci U S A. 2021;118(24).
112. Fu YZ, Wang SY, Zheng ZQ, Yi H, Li WW, Xu ZS, et al. SARS-CoV-2 membrane glycoprotein M antagonizes the MAVS-mediated innate antiviral response. Cell Mol Immunol. 2021;18(3):613-20.
113. Zheng Y, Zhuang MW, Han L, Zhang J, Nan ML, Zhan P, et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) membrane (M) protein inhibits type I and III interferon production by targeting RIG-I/MDA-5 signaling. Signal Transduct Target Ther. 2020;5(1):299.
114. Chen K, Xiao F, Hu D, Ge W, Tian M, Wang W, et al. SARS-CoV-2 Nucleocapsid Protein Interacts with RIG-I and Represses RIG-Mediated IFN-beta Production. Viruses. 2020;13(1).
115. Oh SJ, Shin OS. SARS-CoV-2 Nucleocapsid Protein Targets RIG-I-Like Receptor Pathways to Inhibit the Induction of Interferon Response. Cells. 2021;10(3).
116. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b Is a Potent Interferon Antagonist Whose Activity Is Increased by a Naturally Occurring Elongation Variant. Cell Rep. 2020;32(12):108185.
117. Miorin L, Kehrer T, Sanchez-Aparicio MT, Zhang K, Cohen P, Patel RS, et al. SARS-CoV-2 Orf6 hijacks Nup98 to block STAT nuclear import and antagonize interferon signaling. Proc Natl Acad Sci U S A. 2020;117(45):28344-54.
118. Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020;11(1):3810.
119. Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020;286:198074.
120. Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, et al. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021;34(7):108761.
121. Han L, Zhuang MW, Deng J, Zheng Y, Zhang J, Nan ML, et al. SARS-CoV-2 ORF9b antagonizes type I and III interferons by targeting multiple components of the RIG-I/MDA-5-MAVS, TLR3-TRIF, and cGAS-STING signaling pathways. J Med Virol. 2021;93(9):5376-89.
122. Cortese M, Lee JY, Cerikan B, Neufeldt CJ, Oorschot VMJ, Kohrer S, et al. Integrative Imaging Reveals SARS-CoV-2-Induced Reshaping of Subcellular Morphologies. Cell Host Microbe. 2020;28(6):853-66 e5.
123. Zhang Y, Chen Y, Li Y, Huang F, Luo B, Yuan Y, et al. The ORF8 protein of SARS-CoV-2 mediates immune evasion through down-regulating MHC-Iota. Proc Natl Acad Sci U S A. 2021;118(23).
124. V'Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS-CoV-2. Nat Rev Microbiol. 2021;19(3):155-70.
125. Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell. 2020;183(6):1520-35 e14.
126. Zhang Z, Zheng Y, Niu Z, Zhang B, Wang C, Yao X, et al. SARS-CoV-2 spike protein dictates syncytium-mediated lymphocyte elimination. Cell Death Differ. 2021;28(9):2765-77.
127. Buchrieser J, Dufloo J, Hubert M, Monel B, Planas D, Rajah MM, et al. Syncytia formation by SARS-CoV-2-infected cells. EMBO J. 2020;39(23):e106267.
128. Rajah MM, Bernier A, Buchrieser J, Schwartz O. The Mechanism and Consequences of SARS-CoV-2 Spike-Mediated Fusion and Syncytia Formation. J Mol Biol. 2021:167280.
129. Harvey WT, Carabelli AM, Jackson B, Gupta RK, Thomson EC, Harrison EM, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19(7):409-24.
130. Thorne LG, Bouhaddou M, Reuschl AK, Zuliani-Alvarez L, Polacco B, Pelin A, et al. Evolution of enhanced innate immune evasion by the SARS-CoV-2 B.1.1.7 UK variant. bioRxiv. 2021.
131. Choy KT, Wong AY, Kaewpreedee P, Sia SF, Chen D, Hui KPY, et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antiviral Res. 2020;178:104786.
132. Magro G. COVID-19: Review on latest available drugs and therapies against SARS-CoV-2. Coagulation and inflammation cross-talking. Virus Res. 2020;286:198070.
133. Ali N. Recent Evidence and Possible Therapy against COVID-19-Mediated Hepatic Dysfunction. J Environ Pathol Toxicol Oncol. 2021;40(4):33-41.
134. Te Velthuis AJW, Zubkova TG, Shaw M, Mehle A, Boltz D, Gmeinwieser N, et al. Enisamium Reduces Influenza Virus Shedding and Improves Patient Recovery by Inhibiting Viral RNA Polymerase Activity. Antimicrob Agents Chemother. 2021;65(4).
135. Boltz D, Peng X, Muzzio M, Dash P, Thomas PG, Margitich V. Activity of enisamium, an isonicotinic acid derivative, against influenza viruses in differentiated normal human bronchial epithelial cells. Antivir Chem Chemother. 2018;26:2040206618811416.
136. Holubovska O, Bojkova D, Elli S, Bechtel M, Boltz D, Muzzio M, et al. Enisamium is an inhibitor of the SARS-CoV-2 RNA polymerase and shows improvement of recovery in COVID-19 patients in an interim analysis of a clinical trial. medRxiv. 2021.
137. Singh AK, Singh A, Singh R, Misra A. Molnupiravir in COVID-19: A systematic review of literature. Diabetes Metab Syndr. 2021;15(6):102329.
138. Mahase E. Covid-19: UK becomes first country to authorise antiviral molnupiravir. BMJ. 2021;375:n2697.
139. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, Kovalchuk E, Gonzalez A, Delos Reyes V, et al. Molnupiravir for Oral Treatment of Covid-19 in Nonhospitalized Patients. N Engl J Med. 2021.
140. Holman W, Holman W, McIntosh S, Painter W, Painter G, Bush J, et al. Accelerated first-in-human clinical trial of EIDD-2801/MK-4482 (molnupiravir), a ribonucleoside analog with potent antiviral activity against SARS-CoV-2. Trials. 2021;22(1):561.
141. Mahase E. Covid-19: Pfizer's paxlovid is 89% effective in patients at risk of serious illness, company reports. BMJ. 2021;375:n2713.
142. Merck. https://www.merck.com/news/merck-and-ridgebacks-investigational-oral-antiviral-molnupiravir-reduced-the-risk-of-hospitalization-or-death-by-approximately-50-percent-compared-to-placebo-for-patients-with-mild-or-moderat/. 2021.
143. Yoshida M, Worlock KB, Huang N, Lindeboom RGH, Butler CR, Kumasaka N, et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature. 2021.
144. Samanta M, Iwakiri D, Kanda T, Imaizumi T, Takada K. EB virus-encoded RNAs are recognized by RIG-I and activate signaling to induce type I IFN. EMBO J. 2006;25(18):4207-14.
145. Zhao Y, Ye X, Dunker W, Song Y, Karijolich J. RIG-I like receptor sensing of host RNAs facilitates the cell-intrinsic immune response to KSHV infection. Nat Commun. 2018;9(1):4841.
146. Chiang JJ, Sparrer KMJ, van Gent M, Lassig C, Huang T, Osterrieder N, et al. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat Immunol. 2018;19(1):53-62.
147. Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, et al. Antiviral immunity via RIG-I-mediated recognition of RNA bearing 5'-diphosphates. Nature. 2014;514(7522):372-5.
148. Dou Y, Yim HC, Kirkwood CD, Williams BR, Sadler AJ. The innate immune receptor MDA5 limits rotavirus infection but promotes cell death and pancreatic inflammation. EMBO J. 2017;36(18):2742-57.
149. Zust R, Cervantes-Barragan L, Habjan M, Maier R, Neuman BW, Ziebuhr J, et al. Ribose 2'-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat Immunol. 2011;12(2):137-43.
150. Zalinger ZB, Elliott R, Rose KM, Weiss SR. MDA5 Is Critical to Host Defense during Infection with Murine Coronavirus. J Virol. 2015;89(24):12330-40.
151. Kouwaki T, Nishimura T, Wang G, Oshiumi H. RIG-I-Like Receptor-Mediated Recognition of Viral Genomic RNA of Severe Acute Respiratory Syndrome Coronavirus-2 and Viral Escape From the Host Innate Immune Responses. Front Immunol. 2021;12:700926.
152. Cardenas WB, Loo YM, Gale M, Jr., Hartman AL, Kimberlin CR, Martinez-Sobrido L, et al. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J Virol. 2006;80(11):5168-78.
153. Dutta M, Robertson SJ, Okumura A, Scott DP, Chang J, Weiss JM, et al. A Systems Approach Reveals MAVS Signaling in Myeloid Cells as Critical for Resistance to Ebola Virus in Murine Models of Infection. Cell Rep. 2017;18(3):816-29.
154. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, et al. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J Virol. 2011;85(3):1224-36.
155. Ringeard M, Marchand V, Decroly E, Motorin Y, Bennasser Y. FTSJ3 is an RNA 2'-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature. 2019;565(7740):500-4.
156. Lu HL, Liao F. Melanoma differentiation-associated gene 5 senses hepatitis B virus and activates innate immune signaling to suppress virus replication. J Immunol. 2013;191(6):3264-76.
157. Sato S, Li K, Kameyama T, Hayashi T, Ishida Y, Murakami S, et al. The RNA sensor RIG-I dually functions as an innate sensor and direct antiviral factor for hepatitis B virus. Immunity. 2015;42(1):123-32.
158. Zhou L, He R, Fang P, Li M, Yu H, Wang Q, et al. Hepatitis B virus rigs the cellular metabolome to avoid innate immune recognition. Nat Commun. 2021;12(1):98.