Abstract
Recent decades have witnessed the emergence of many viral diseases some of which have resulted in epidemics and global pandemics. Design of drugs and vaccines for such diseases is expensive and time consuming. In this article we propose a new computer-assisted method for the fast design of a new class of vaccines, the peptide vaccines, based on the genetic sequences of the pathogens. It is expected that this approach will assist in fighting emerging pathogens.
Keywords
Bioinformatics, Vaccines, COVID-19
Introduction
The currently ongoing coronavirus pandemic, the SARSCOV- 2, interchangeably referred to as the COVID-19 infection, has in a short span of time altered the ways and means of almost all of mankind. So strong has been its effect that all human activity ceased in one way or another for a considerable time, led to significant loss of life and economic drain of untold proportions such that there are open debates on whether the world will forever now change from the way we were used to [1]. Just like in the case of the several epidemics that have plagued human society in this 21st century, such as severe acute respiratory syndrome coronavirus (SARS-CoV) from 2002 to 2003 [2], the Middle East respiratory syndrome coronavirus (MERS-CoV) of 2012 [3], and H1N1 influenza in 2009 [4], there are no drugs or vaccines available at this time for the SARS-CoV-2, but considering its impact efforts are under way in more than 100 labs worldwide to develop a vaccine with great urgency.
The fatality ratio for the new virus is not as high as for the SARS-CoV, but its spread and death toll make for lessons in epidemics and pandemics which are occurring with increasing frequency in the last few decades. The genetic makeup of this virus is like the other coronaviruses; this virus also seems to be of zoonotic origin, possibly bats, that has made the jump to infect humans [5,6]. The SARSCoV- 2 viral genome is a single-stranded positive sense RNA polyprotein that is among some of the largest viral genomes, up to around 32 kbp in length [7] belonging to the genus Betacoronavirus within the family Coronaviridae [8]. The SARS-CoV-2 genome contains the usual groups of structural and non-structural genes. At the virion level, the trimeric spike (S) glycoproteins are surface situated and mediate entry into host cells by binding to the human angiotensin converting enzyme 2 (ACE2) protein as a host receptor [9,10]. The S protein has two subunits: S1 that mediates cell attachment, and S2 that mediates the fusion of the viral and cellular membrane [11]. The SARS-CoV-2 virus is a singularly virulent specimen that infects silently and shows no symptoms for the first week or two while actively spreading through droplet infection resulting in rapid human-to-human transmission; there have been recent evidence of airborne transmission too [12]. After first reports of a new viral disease in Wuhan, China in December 2019, the virus spread across the whole world in a matter of weeks. The effects of the disease differ by age, gender, race and comorbidity [1]. Clinical symptoms include dry cough, high fever, respiratory tract symptoms, diarrhea, among others, although a significant percentage of infected persons remain asymptomatic [13]. Potential risk factors for COVID-19 severity of patients are older age, high Sequential Organ Failure Assessment (SOFA) scores, and d-dimer concentration greater than 1 μg/mL [14].
COVID-19 clinical and pathological profiles are different from the Zika virus pandemic where the more significant clinical damages were most often confined to pregnant women in the Western hemisphere [15]. A cause of singular consternation at the time of its epidemic, there were no drugs or vaccines available then and that situation persists till today; there are a number of viruses such as dengue, chikungunya, Zika and others where development of vaccines has faced unexpected hurdles and only recourse is symptom relief when these viral diseases recur. Unfortunately, the number of new viruses or mutated forms of old viruses continue to multiply at ever greater frequency leading to further anxiety and consequences.
Traditionally, anti-viral vaccines have been of the weakened or inactivated form or VLPs (virus like particles) [16] and typically takes several years and billions of dollars to develop [17]; then also, because viruses, especially RNA viruses, mutate very rapidly, the vaccines would not be sustainable for long, and therefore are usually commercially unviable. Further, all vaccines developed to date are considered universal, i.e. applicable to all humans, without any community specificity or tailored to individual needs. That such needs can arise is typified by the extreme case of a patient suffering from a fatal but rare neurodegenerative disease for whose treatment alone a new drug, milasen, was manufactured [18]. Also, consider that these viruses have become endemic to different population groups; see Vracko et al. [19] where the Zika glycoprotein is clearly clustered in geographically different groups. A universal vaccine may not always cater with same efficiency to all these varieties of the Zika virus, and the varieties are expected to grow with mutations over time.
Thus, there is clearly a need to think anew. With growing knowledge and understanding of the human immune system, immunogenetics and significant developments in immunoinformatics, bioinformatics, genomics, information technology (IT) and allied subjects, there have been efforts to focus attention on the function of the anti-viral vaccines and how they work. This has led to the concepts of “reverse vaccinology” [20] and “vaccinomics” [21,22] which allows for a more focused approach towards the development of vaccines based on specific surface exposed proteins of the virus. That approach can enable rapid development of vaccines to combat an explosive epidemic [23,24] and make small adjustments to the vaccine to respond better to communities and even individuals if the situation warrants [25], far removed from the “one size fits all” concept of the traditional vaccines.
Peptide Vaccines
Peptide vaccines are one of the results of new paradigm where targeted vaccines are indicated. Although it was propounded many years ago against canine parovirus [26], malaria [27] and swine fever virus [28] in animals, the subject lay dormant until applications and trials on human patients in recent years brought it into significance. The trials on cancerous tumors gave very encouraging results leading Singluff [29] to remark that peptide vaccines will have 100% success rate in such cases. Brossart et al. [30] determined that patients with advanced breast and ovarian cancers can be treated with peptide vaccines derived from MUC1 ( mucin1, one of the several mucus proteins); Ludewig et al. [31] found anti-tumor immune response on administration of peptide antigen-based vaccine against lymphocytic choriomeningitis virus, while Liao et al. [32] found mice could gain strong cell-mediated immunity and be protected from tumor growth when injected with human papilloma virus E5 peptide vaccine along with a suitable adjuvant, among other such examples.
The reports for communicable diseases are still preliminary. There are many peptide vaccine trials listed in ClinicalTrials.com website, but mostly for cancers; the remaining are for phase trials against various viruses such as dengue, measles, Zika, etc. The National Institute of Allergy and Infectious Diseases (NIAID) of the USA had taken up the case for a vaccine of the Zika virus with some urgency [33]; at the present time the CoVID-19 virus has become the predominant concern [34]. A 9-peptide combination vaccine against influenza A and B was under trial by BiondVax Pharmaceuticals Ltd. [35], Islam et al. [36] determined a set of conserved regions in the chikungunya glycoprotein E2 by sequence alignment for consideration as epitopes for peptide vaccine design, Chakraborty et al. [37] analysed sequences of all four dengue virus types glycoprotein to get highly conserved surface accessible segments with high antigenicity and low hydrophobicity to be tried as peptide vaccine candidates. These were all in silico analyses, wet lab experiments were awaited for different phase trials.
Peptide vaccines thus show good promise as anti-viral prophylactics. Such vaccines have some natural advantages. They are well-targeted to impact only the designated viral segments, they are selected to pose no auto-immune threats, being a chemical product they are easily manufactured and purity assured, storage and transportation are much more easily managed like an industrial product without having to use very low-temperature refrigeration as for current vaccines, administration to a community is no different from the current practice, and several others [38,39].
However, despite good and encouraging results in various peptide vaccine trial runs, no such vaccines have yet been licensed for human use. Some of the reasons for the delay in licensing procedure are the issues related to its function [40]. It has been observed that the burden of excess T-cell leukocyte production through the peptide vaccine route have adverse effect on the host body, careful control of which becomes quite critical. Then also, the peptide vaccines themselves are not capable of generating adequate antibodies to counter a pathogen’s onslaught; they need support using adjuvants, of which there are now only a few types in practice. Stability of the peptide in vivo is another issue: Short peptides can fold quite easily and not maintain their required conformations thus requiring other measures to induce the desired structure [41].
Tools for Peptide Vaccine Design
We have determined using computational methods many peptides that could be used as anti-viral vaccines for different viruses [42-45]. While different research groups have different approaches for designing peptide vaccines (see e.g., Ref. [46]), we have added one additional feature in our standardized protocol, viz., that the peptides we classify as possible vaccine targets should, in addition to being well exposed to the solvent, also be evolutionarily as much conserved as possible to enable a longer shelf life for the vaccines than otherwise. This requires careful identification of distinct sequences so that preponderance of one type of sequence does not overwhelm the search for variety. While most molecular biologists would perform an alignment based NCBI BLAST analysis [47] for the purpose, such procedures take a long time to conclude, and are extremely resource hungry such that only a few tens or hundreds of sequences can be considered at a time. Alignment-free analysis have recently been proposed that are less resource hungry, of which graphical representation and numerical characterization have drawn considerable attention [48]. One model that we have adopted [49], plots a DNA/RNA sequence in a 2D graph where the four axes are identified with the four nucleotides in some preordained manner such as, e.g., -ve x-axis with adenine, positive y-axis with cytosine, +ve x-axis with guanine and -ve y-axis with thymine/uracil. Thus, given a DNA/ RNA sequence one has to take a step in the appropriate direction starting from the origin for the beginning of the sequence for each base until the end which plots out a graph that visually shows the distribution of bases along the sequence. Numerically, one may compute the weighted average of x,y co-ordinates, μx, μy, and then compute a graph radius gR [50]
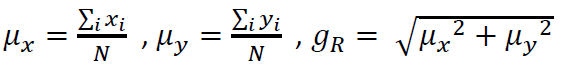

The gR are found to be specific to each sequence and any change in the sequence reflects in a change in the gR thus making it a good descriptor of the sequence and a great filter to weed out identical sequences [51].
Likewise, one can define a descriptor for protein sequences too, except this time there are 20 amino acids so we define a 20D cartesian co-ordinate system and plot a graph in the abstract space using the same technique as for nucleotide sequences [52]. The graph radius, pR, this time defined as
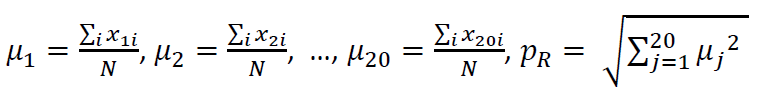
which is also the sequence descriptor, pR, is found to differ if there is any change in the sequence and thus can be used to filter out identical sequences. Another graphical representation is a 2D polar plot [53] where the amino acids are assigned along radii 18 degrees apart. Plotting a sequence in this graph represents amino acid distribution along the sequence and therefore allows comparison between protein sequences that have their amino acid distribution altered by mutations. Here also one can define a graph radius, qR, analogously to the pR and is seen to have similar properties like identical qR between two sequences implying sequence identity with no mutational changes.
GSWM and 2D polygon representation
Of particular interest is the GSWM technique. This is used to determine conserved segments in a family of protein sequences. Thus, we take a window size of 12 amino acids (selected as a compromise candidate for MHC Class I and Class II groove capacity [54] and compute the pR of the segment. Next, we move along the sequence by one step and compute the pR for the new window, continuing likewise till the end of the sequence. We can then do the same exercise for the next protein sequence and the following ones and line up the pR values for each window for each sequence amino acid number-wise. Scanning the pR values in each window we can determine how many different values there are. This is a measure of protein variability (PV), the lower the number the more conserved is the segment. This graphical sliding widow method (GSWM) has been of immense advantage in determining segment-wise conservancy of protein sequences.
For our objective of identifying protein segments that are highly surface exposed (high ASA) and well conserved (low PV), we consider a mathematical framework where at each amino acid position in the protein sequence we compute the area of a triangle whose sides are of length ASA, 1/PV and ASA-PV (see details in Ref [55,56]). Clearly, the bigger the area the closer we are to our objective. For linear epitopes we take contiguous large area triangles in order of size to form the peptides desired. Choosing the first ten or 20 of such peptides we can then perform BLAST analysis with human proteins to ensure no auto immune threats and then short-list the top 10 best candidates for probable wet lab experiments.
The Peptide Design Protocol
Using several of these tools we had analyzed different viruses to determine the most appropriate epitopes for wet-lab analysis. However, the methods we had employed required manual intervention at several stages (see Ref [57]). Recently we have updated our analytical techniques such that much of the work is computerized and the protocol changed appropriately. Thus, the new protocol follows the following steps to design a vaccine candidate for a particular viral disease:
- Determine the best surface protein of the virus for vaccine targeting and collect as much sequence data as possible;
- Use the graphical methods to retain only unique sequences;
- Compute or use suitable web-based servers to get the average solvent accessibility (ASA) of the viral sequence residues;
- Use the GSWM to compute protein variability (PV);
- Compute and list the best combinations using the 2D Polygon method;
- Determine the epitope potential of the selected segments through web-based servers like IEDB (Immune Epitope DataBase Analysis Resource)
- Check with BLAST for auto-immune threats, if any;
- Create the final short list of best epitopes for vaccine design.
If 3D structure information on the targeted protein is available, it is preferable to check that indeed the peptides determined through the above process are surface accessible. Such accessibility may be curtailed to a smaller or greater extent from neighboring proteins, especially in multimeric proteins such as tetrameric in case of influenza neuraminidase, or trimeric for SARS-CoV-2 spike glycoprotein. In case there are substantial overlaps, the recommendation of peptide targets may need to be modified. There are several other issues that may arise while following the protocols; some of these are described in Nandy et al. [57], practical considerations may help decide on others. Using this protocol we have reworked the vaccine targets for previous investigations and found that they generally conform quite well and also added a few more candidates to the peptide vaccines short list [58]; an early prediction of peptide vaccine targets for the CoVID-19 virus was also published [56].
Discussions
It is now well understood that peptide vaccines targeted at specific regions of the surface proteins of the virions can augment the human immune system’s ability to destroy invading pathogens. While it has been observed that the intensity of antibody generation through the peptide vaccine route is not as abundant as in the case of traditional vaccines, addition of a carrier protein and an adjuvant bolsters the immunity development and response system. Although no results can be guaranteed, however, the peptide vaccine route remains the fastest response that can be contemplated against a rampaging pandemic, except for lucky breaks like the Ebola epidemic where a repurposed anti-viral could be used to stem the flow, or the world gears up to produce a vaccine in record time as is being tried out for the COVID-19. Note that 4-5 years after the Zika virus pandemic, there is still a lack of viable vaccines of any type [33].
One of the major issues that concern immune development process is the mutational effects on viruses. Viruses are generally of the RNA type where the replication machinery does not include any error correction process but must depend upon the host replication systems for eventual growth and replication. RNA viruses therefore are found to mutate at 10-4 to 10-5 times per nucleotide per replication, much faster than DNA varieties; for a typical influenza genome with about 10k bases, there can be one mutation per replication. In the case of the Zika virus it has been found that the effective mutation change is at a rate of 0.12% to 0.25% of the polyprotein length of about 10300 nt per year, which is quite a rapid rate [59]. The COVID-19 that appeared in Wuhan, China in December 2019, was thought initially to have a slow mutation rate, but mutation tracking by Wrobel et al. [60] in the spike glycoprotein showed several mutations with one becoming the dominant strain in Europe, also remarked by Korber et al. [61], while recent additions to the SARS-COV-2 database in NCBI GenBank shows that there is a rapid accumulation of mutant varieties [62], although it is too early yet to arrive at any rate estimate of the mutations taking place. It is interesting to note that this has a close parallel with the Zika virus history where too the early years saw slow mutational changes which later burgeoned by at least an order of magnitude [63].
Error-prone replication that alters the virus to mutate to new forms effectively allows the virus to escape immune surveillance. For viruses like the flu with a high mutation rate, this requires new vaccine design every year based on observations of the viral sequences of the current year, which may not always give satisfactory results; the flu vaccine for 2019 fell quite short of expectations in the USA [64]. The strategies for vaccine design for such highly mutable viruses like the ‘flu or HIV1 have thus focused on well conserved regions and epitopes.
Traditional weakened or inactivated vaccines can address all available epitopes, irrespective of their efficacy, on the virion, whereas peptide vaccines are designed to operate against only one or a few epitopes. Since some epitopes can be more predominant than others, it is important that the immunodominance factor is considered when designing peptide vaccines. This can be taken care of through the concept of multivalent peptide vaccines where several peptide vaccines that may also include the immunodominant region are bunched together in a carrier protein for administration to the target host population. Such multivalent vaccines may include more than one epitope region from a single virus strain or epitopes from other strains of the same virus, or from genetically separate types of the same viral family [65]. While not a peptide vaccine, the Gardasil vaccine marketed by Merck & Co against 9 types of human papillomavirus is one of the few multivalent vaccines that uses VLP technology; elsewhere Joura et al. [66] designed 9-valent HPV vaccine against certain infections in women. The dengue virus with 4 genetically different types, but with the critical antibody-dependent enhancement possibilities, remains a challenging target for single or a multivalent vaccine.
While CoVID-19, unlike previous epidemics, is a totally new experience for mankind, more epidemics are likely to arise than not. Global warming is permitting tropical viral vectors to spread into wider areas [67] and new reservoirs of viruses are being discovered (e.g., [68]). Like the AIDs and Zika, Nipha and COVID-19, epidemics from new zoonotic viruses against which we have no prior immunity are likely to arise with increasing frequency from conflicts of interest arising from a burgeoning human population looking for land to expand and a shrinking forest and ecological cover for survival of wild animals [69]. In such a scenario, viruses that are now confined to animal eco-systems can make the jump to humans in close encounters and spawn new viral diseases. Such prospects require rapid response route, where automated systems, robust computational infrastructure and rational design of vaccines will have to take the lead [23]. Elsewhere we have proposed spanning the globe with peptide vaccine factories that can take the lead molecule from designated laboratories and tailor them to their communities for maximum effect [25] within short time scales.
Thus, peptide vaccines pose an intriguing possibility of rapid development and deployment which is so much a necessity in the face of surging epidemics and pandemics, and the increasing frequency of new emergent viruses. Surveillance and epidemiological studies need to be pursued at high pitch with ready technological infrastructure lest we lose the keys to knowledge of possible viral intrusions learnt at great cost in the Zika virus pandemic [70,71]. Several issues remain to be solved, but the prospects of such peptide vaccines are to be anticipated with lots of hope for developing countries where per capita income is low and infrastructure to support the environment required for the currently available traditional type vaccines is relatively scarce. Given the zeal with which laboratories around the world have launched into developing a vaccine against the COVID-19, and the successes that have been observed against cancer tumors, prospects of licensure for some peptide vaccines may not be very far away.
Author Contributions
AN conceived of the project and wrote the manuscript, SM was involved with the general introduction and SCB advised and critically reviewed the project and manuscript.
References
2. Hui DS, Zumla A. Severe acute respiratory syndrome: historical, epidemiologic, and clinical features. Infectious Disease Clinics. 2019 Dec 1;33(4):869-89.
3. Azhar EI, Hui DS, Memish ZA, Drosten C, Zumla A. The middle east respiratory syndrome (MERS). Infectious Disease Clinics. 2019 Dec 1;33(4):891-905.
4. Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, et al. Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study. The Lancet Infectious Diseases. 2012 Sep 1;12(9):687-95.
5. Cascella M, Rajnik M, Cuomo A, Dulebohn SC, Di Napoli R. Features, evaluation and treatment coronavirus (COVID-19). InStatpearls [internet]2020 Mar 8. StatPearls Publishing.
6. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nature Medicine. 2020 Apr;26(4):450-2.
7. Cui J, Li F, Shi ZL. Origin and evolution of pathogenic coronaviruses. Nature Reviews Microbiology. 2019 Mar;17(3):181-92.
8. Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology. 2020;5(4):536.
9. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 Mar;579(7798):270-3.
10. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003 Nov;426(6965):450-4.
11. Walls AC, Xiong X, Park YJ, Tortorici MA, Snijder J, Quispe J, Cameroni E, Gopal R, Dai M, Lanzavecchia A, Zambon M. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell. 2019 Feb 21;176(5):1026-39.
12. Zhang R, Li Y, Zhang AL, Wang Y, Molina MJ. Identifying airborne transmission as the dominant route for the spread of COVID-19. Proceedings of the National Academy of Sciences. 2020 Jun 11;117:14857-14863.
13. World Health Organization, Coronavirus disease (COVID-2019) situation reports. https://www.who.int/ emergencies/diseases/novel-coronavirus-2019/situationreports/. Accessed July 16, 2020.
14. Zhou F, Yu T, Du R, Fan G, Liu Y, Liu Z, et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. The Lancet. 2020 Mar 11; 395:1054-1062.
15. Nandy A, Basak SC. The Epidemic that Shook the World-The Zika Virus Rampage. Exploratory Research and Hypothesis in Medicine. 2017 Sep 25;2(3):43-56.
16. Garg H, Mehmetoglu-Gurbuz T, Joshi A. Virus Like Particles (VLP) as multivalent vaccine candidate against Chikungunya, Japanese Encephalitis, Yellow Fever and Zika Virus. Scientific Reports. 2020 Mar 4;10(1):1-3.
17. Chit A, Parker J, Halperin SA, Papadimitropoulos M, Krahn M, Grootendorst P. Toward more specific and transparent research and development costs: The case of seasonal influenza vaccines. Vaccine. 2014 May 30;32(26):3336-40.
18. Kim J, Hu C, Moufawad El Achkar C, Black LE, Douville J, Larson A, et al. Patient-customized oligonucleotide therapy for a rare genetic disease. New England Journal of Medicine. 2019 Oct 24;381(17):1644-52.
19. Vracko M, Basak SC, Sen D, Nandy A. Clustering of Zika viruses originating from different geographical regions using computational sequence descriptors. Current computer-aided drug design. 2019 Dec 25.
20. Rappuoli R. Reverse vaccinology, a genome-based approach to vaccine development. Vaccine. 2001 Mar 21;19(17-19):2688-91.
21. Poland GA, Kennedy RB, Ovsyannikova IG. Vaccinomics and personalized vaccinology: is science leading us toward a new path of directed vaccine development and discovery?. PLoS Pathogen. 2011 Dec 29;7(12):e1002344.
22. Poland GA, Whitaker JA, Poland CM, Ovsyannikova IG, Kennedy RB. Vaccinology in the third millennium: scientific and social challenges. Current Opinion in Virology. 2016 Apr 1;17:116-25.
23. Nandy A, Basak SC. Viral epidemics and vaccine preparedness. Journal of MPE Molecular Pathological Epidemiology. 2017;2:S1.
24. Nandy A, Dey S, Roy P, Basak SC. Epidemics and peptide vaccine response: A brief review. Current Topics in Medicinal Chemistry. 2018 Oct 1;18(26):2202-8.
25. Nandy A, Basak SC. Towards Personalized Vaccines- Tailoring Peptide Vaccines to Demographic Groups and Individuals. Med One. 2019 Dec 23;4(6).
26. Langeveld JP, Casal JI, Osterhaus AD, Cortes E, De Swart R, Vela C, et al. First peptide vaccine providing protection against viral infection in the target animal: studies of canine parvovirus in dogs. Journal of Virology. 1994 Jul 1;68(7):4506-13.
27. Wang R, Charoenvit Y, Corradin G, Porrozzi R, Hunter RL, Glenn G, et al. Induction of protective polyclonal antibodies by immunization with a Plasmodium yoelii circumsporozoite protein multiple antigen peptide vaccine. The Journal of Immunology. 1995 Mar 15;154(6):2784-93.
28. Mons- M, Tarradas J, de la Torre BG, Sobrino F, Ganges L, Andreu D. Peptide vaccine candidates against classical swine fever virus: T cell and neutralizing antibody responses of dendrimers displaying E2 and NS2-3 epitopes. Journal of Peptide Science. 2011 Jan;17(1):24-31.
29. Slingluff Jr CL. The present and future of peptide vaccines for cancer: single or multiple, long or short, alone or in combination?. Cancer Journal (Sudbury, Mass.). 2011 Sep;17(5):343.
30. Brossart P, Wirths S, Stuhler G, Reichardt VL, Kanz L, Brugger W. Induction of cytotoxic T-lymphocyte responses in vivo after vaccinations with peptide-pulsed dendritic cells. Blood, The Journal of the American Society of Hematology. 2000 Nov 1;96(9):3102-8.
31. Ludewig B, Barchiesi F, Pericin M, Zinkernagel RM, Hengartner H, Schwendener RA. In vivo antigen loading and activation of dendritic cells via a liposomal peptide vaccine mediates protective antiviral and anti-tumour immunity. Vaccine. 2000 Aug 15;19(1):23-32.
32. Liao SJ, Deng DR, Zeng D, Zhang L, Hu XJ, Zhang WN, et al. HPV16 E5 peptide vaccine in treatment of cervical cancer in vitro and in vivo. Journal of Huazhong University of Science and Technology [Medical Sciences]. 2013 Oct 1;33(5):735-42.
33. NIAID 2020. NIH Begins Testing Investigational Zika Vaccine in Humans. https://www.niaid.nih.gov/newsevents/ nih-begins-testing-investigational-zika-vaccinehumans. Last reviewed July 16, 2020. Last accessed July 17, 2020.
34. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, et al. An mRNA vaccine against SARS-CoV-2-preliminary report. New England Journal of Medicine. 2020 Jul 14.
35. BiondVax. Assess the Safety and Immunogenicity of M-001 as A Standalone Influenza Vaccine and as A H5N1 Vaccine Primer in Adults. ClinicalTrials.gov Identifier: NCT02691130. Last update February 2018. Last accessed July 17, 2020.
36. Islam R, Sakib MS, Zaman A. A computational assay to design an epitope-based peptide vaccine against chikungunya virus. Future Virology. 2012 Oct;7(10):1029- 42.
37. Chakraborty S, Chakravorty R, Ahmed M, Rahman A, Waise TM, Hassan F, et al. A computational approach for identification of epitopes in dengue virus envelope protein: a step towards designing a universal dengue vaccine targeting endemic regions. In silico Biology. 2010 Jan 1;10(5, 6):235-46.
38. Purcell AW, McCluskey J, Rossjohn J. More than one reason to rethink the use of peptides in vaccine design. Nature Reviews Drug Discovery. 2007 May;6(5):404-14.
39. Moisa, A.A.; Kolesanova, E.F. Synthetic peptide vaccines. In Insight and Control of Infectious Disease in Global Scenario; Roy, P., Ed.; ISBN: 978-953-51-0319-6; InTech: Rijeka, Croatia, 2012.
40. Celis E. Getting peptide vaccines to work: just a matter of quality control?. The Journal of Clinical Investigation. 2002 Dec 15;110(12):1765-8.
41. Skwarczynski M, Toth I. Peptide-based synthetic vaccines. Chemical science. 2016;7(2):842-54.
42. Ghosh A, Nandy A, Nandy P. Computational analysis and determination of a highly conserved surface exposed segment in H5N1 avian flu and H1N1 swine flu neuraminidase. BMC Structural Biology. 2010 Dec;10(1):1- 10.
43. Ghosh A, Chattopadhyay S, Chawla-Sarkar M, Nandy P, Nandy A. In silico study of rotavirus VP7 surface accessible conserved regions for antiviral drug/vaccine design. PLoS One. 2012 Jul 26;7(7):e40749.
44. Dey S, Nandy A, Basak SC, Nandy P, Das S. A bioinformatics approach to designing a Zika virus vaccine. Computational Biology and Chemistry. 2017 Jun 1;68:143- 52.
45. Sarkar T, Das S, De A, Nandy P, Chattopadhyay S, Chawla-Sarkar M, et al. H7N9 influenza outbreak in China 2013: In silico analyses of conserved segments of the hemagglutinin as a basis for the selection of peptide vaccine targets. Computational Biology and Chemistry. 2015 Dec 1;59:8-15.
46. Abdelmageed MI, Abdelmoneim AH, Mustafa MI, Elfadol NM, Murshed NS, Shantier SW, et al. Design of a Multiepitope-Based Peptide Vaccine against the E Protein of Human COVID-19: An Immunoinformatics Approach. BioMed Research International. 2020 Jan 1;2020.
47. NCBI BLAST (2019) https://blast.ncbi.nlm.nih.gov/ Blast.cgi
48. Nandy A, Harle M, Basak SC. Mathematical descriptors of DNA sequences: development and applications. Arkivoc. 2006 Jan 1;9(2006):211-38.
49. Nandy A. A new graphical representation and analysis of DNA sequence structure: I. Methodology and application to globin genes. Current Science. 1994 Feb 25:309-14.
50. Raychaudhury C, Nandy A. Indexing scheme and similarity measures for macromolecular sequences. Journal of Chemical Information and Computer Sciences. 1999 Mar 22;39(2):243-7.
51. Nandy A, Nandy P. On the uniqueness of quantitative DNA difference descriptors in 2D graphical representation models. Chemical Physics Letters. 2003 Jan 14;368(1- 2):102-7.
52. Nandy A, Ghosh A, Nandy P. Numerical characterization of protein sequences and application to voltage-gated sodium channel a subunit phylogeny. In silico Biology. 2009 Jan 1;9(3):77-87.
53. Deya T, Biswas S, Chatterjee S, Manna S, Nandye A, Basake SC. 2D Polar Co-ordinate Representation of Amino Acid Sequences With some applications to Ebola virus, SARS and SARS-CoV-2 (COVID-19).
54. Malonis RJ, Lai JR, Vergnolle O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chemical reviews. 2019 Dec 5;120(6):3210-29.
55. Biswas S, Dey T, Chatterjee S, Manna S, Nandy A, Das S, et al. Novel Algorithms for In Silico Peptide Vaccine Design with reference to Ebola Virus. IEEE International Conference on Computer, Electrical and Communication Engineering (ICCECE 2020), Kolkata, India.
56. Biswas S, Chatterjee S, Dey T, Dey S, Manna S, Nandy A, et al. In Silico Approach for Peptide Vaccine Design for CoVID 19. MOL2NET 2020, International Conference on Multidisciplinary Sciences, 6th edition session, USINEWS-04: US-IN-EU Worldwide Science Workshop Series, UMN, Duluth, USA.
57. Nandy A, Manna S, Basak SC. Computational Methodology for Peptide Vaccine Design for Zika Virus: A Bioinformatics Approach. InImmunoinformatics 2020 (pp. 17-30). Humana, New York, NY.
58. Biswas S, Chatterjee S, Dey T, Dey S, Manna S, Nandy A, et al. In Silico Approach for Peptide Vaccine Design for CoVID 19.
59. Logan IS. ZIKA-How fast does this virus mutate?. Zoological Research. 2016 Mar 18;37(2):110.
60. Wrobel AG, Benton DJ, Xu P, Roustan C, Martin SR, Rosenthal PB, et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nature Structural & Molecular Biology. 2020 Aug;27(8):763-7.
61. Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv preprint doi: https://doi. org/10.1101/2020.04.29.069054.
62. Kupferschmidt K. The pandemic virus is slowly mutating. But is it getting more dangerous. Science. 2020.
63. Yokoyama S, Starmer WT. Possible roles of new mutations shared by Asian and American Zika viruses. Molecular biology and evolution. 2017 Mar 1;34(3):525- 34.
64. CDC MMWR Weekly Report, Feb 21, 2020. Interim Estimates of 2019-20 Seasonal Influenza Vaccine Effectiveness -United States, February 2020.
65. Dey S, De A, Nandy A. Rational design of peptide vaccines against multiple types of human papillomavirus. Cancer Informatics. 2016 Jan;15:CIN-S39071.
66. Joura EA, Giuliano AR, Iversen OE, Bouchard C, Mao C, Mehlsen J, Moreira Jr ED, Ngan Y, Petersen LK, Lazcano-Ponce E, Pitisuttithum P. A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. New England Journal of Medicine. 2015 Feb 19;372(8):711- 23.
67. Shuman EK. Global climate change and infectious diseases. New England Journal of Medicine. 2010 Mar 25;362(12):1061-3.
68. Shi M, Lin XD, Tian JH, Chen LJ, Chen X, Li CX, et al. Redefining the invertebrate RNA virosphere. Nature. 2016 Dec;540(7634):539-43.
69. Editorial. Zoonoses: beyond the human-animal- environment interface. The Lancet 2020;396:1-4.
70. Nandy A, Basak SC. A brief review of computerassisted approaches to rational design of peptide vaccines. International Journal of Molecular Sciences. 2016 May;17(5):666.
71. Basak SC, Bhattacharjee A and Nandy A, Editors. Zika virus Surveillance, Vaccinology and Anti-Zika Drug Discovery - Computer Assisted Strategies to Combat the Menace. Nova Science Publishers, Nova Medicine and Health, New York, New York. 2019.