Keywords
JAKinibs, Bone, Osteoimmunology, Rheumatoid arthritis, Oosteoporosis
Introduction
Cytokine receptors may possess an intrinsic capability for the transduction of signals upon engagement by the respective cytokine ligand [1]. However, if they lack an own intracellular signaling entity, they rely on other signaling machineries. One of the key intracellular signaling molecules mediating cytokine effects on immune cells are Janus kinases (JAKs), which induce gene expression via signal transducer and activator of transcription proteins (STATs). In mammals, four JAK (JAK1, JAK2, JAK3 and Tyk2) and seven STAT proteins (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B and STAT6) are described, which in varying combinations mediate signal transduction of well over fifty cytokines [2,3]. In the following article the role of JAK and STAT will be summarized.
JAK Inhibition in Inflammation
This array of cytokines, which rely on the JAK/STAT machinery, contains proinflammatory mediators, many of which are relevant for inflammatory diseases, such as rheumatoid arthritis (RA). RA is a chronic autoimmune disorder, characterized by a persistent symmetric polyarthritis and synovitis [4,5]. Inflammatory cytokines relevant to the pathogenesis of RA, utilizing the JAK/ STAT pathway, include interleukin-6 (IL-6), IL-7, IL-10, IL-12, IL-15, IL-21, IL-23 and interferons (α, β and γ) [6,7]. Consequently, JAK inhibitors have pursued their way into the clinical practice. Currently, in the USA and the EU three JAK inhibitors have been approved for the treatment of RA, tofacitinib, baricitinib and upadacitinib and a fourth, filgotinib, is about to follow [8-14]. Tofacitinib, conceptually a pan-JAK inhibitor, targets primarily JAK1 and JAK3, while baricitinib inhibits JAK1 and JAK2 [8,13,15-17]. Upadacitinib and filgotinib specifically suppress JAK1 activity [18,19]. McInnes et al. demonstrated differences in the pharmacokinetic profiles of tofacitinib and baricitinib, as well as varying in vitro pharmacology profiles, which highlights their differing substrate specificities [17]. Moreover, a meta-analysis in 2020 compared the efficacy of multiple JAK inhibitors with adalimumab in RA patients on methotrexate background therapy. The authors confirmed that, compared to adalimumab, both baricitinib and tofacitinib exhibit higher probability of achieving ACR20 responses. However, RA patients receiving baricitinib additionally benefited from a significantly higher ACR50 response rate compared to adalimumab therapy [20]. RA patients generally respond well to all JAK inhibitors, which may be based on their inhibition of multiple inflammatory cytokines instead of just one, as the JAK/STAT machinery is shared by several cytokines [21,22].
JAK/STAT Signaling and Bone
Less is known about the role of JAKs on bone homeostasis. However, the role of the JAK/STAT pathways on the immune system inevitably suggests that bone metabolism will also be influenced by JAKs [23]. Preclinical studies have shown that mice knocked out for STAT1 display an increased bone mass [24-26]. Moreover, global STAT3 knockouts are lethal [27]. However, when specifically knocked out in osteoblasts and osteocytes, STAT3 deficient mice develop osteoporosis [28,29]. The clinical relevance of STAT3, as a signaling mediator in bone metabolism, is manifested in the Job’s syndrome, which is the result of a mutation in the STAT3-DNA-binding domain [30-33]. Patients with Job’s syndrome develop, among other symptoms, a bone phenotype: The number of bone-degrading osteoclasts is increased and bone mineral density is reduced. Patients suffer from recurrent fractures, due to increased bone fragility as a consequence of poor bone quality [23,30,32,34-36].
As JAKs and STATs primarily regulate immune function, these molecules and their inhibitors may also affect bone homeostasis during inflammatory diseases [37]. In RA, for instance, local and systemic bone loss develop, which require treatments that not only influence inflammation but also maintain or even restore bone mass [38-40]. There have been indications that JAK inhibitors may pose a beneficial impact on bone metabolism. For instance, Orsolini and colleagues suggested tofacitinib as a mean of limiting bone loss during chronic inflammatory diseases, such as RA and psoriatic arthritis [41]. However, the osteometabolic properties of JAK inhibitors have been systematically studied only recently.
JAK Inhibitors Enhance Bone Formation and Bone Mass
We conducted a preclinical analysis of the impact of JAK inhibition, by tofacitinib and baricitinib, on bone homeostasis in steady-state conditions and during pathological challenge by non-inflammatory bone loss (postmenopausal osteoporosis) and inflammationmediated bone loss (serum transfer arthritis) [42]. In all three models, JAK inhibitors consistently increased bone mass, predominantly of the trabecular compartment. As the numbers of osteoclasts and osteoblasts were unaffected by JAK inhibitor treatment, we suspected a functional influence of JAK inhibition on bone cells.
Therefore, we separately investigated the effect of JAK inhibitors on bone-degrading osteoclasts and boneforming osteoblasts in in vitro cultures. While osteoclasts were not directly affected by JAK inhibitors, JAK inhibition enhanced the mineralization capability and activity of osteoblasts. Whole transcriptome analysis of osteoblasts revealed induction of an osteoanabolic gene signature by JAK inhibitors enhancing the anabolic Wnt signaling pathway (Figure 1). When analyzing JAK/STAT interactions, we found no effect on STAT1-activation but almost complete abrogation of STAT3-activation by JAK inhibitors. Importantly, we explored RA patients for signs of bone repair, after having received JAK inhibitors for at least two years. High-resolution peripheral quantitative computed tomography (HR-pQCT) revealed repair of bone erosions in their hand joints of these patients.
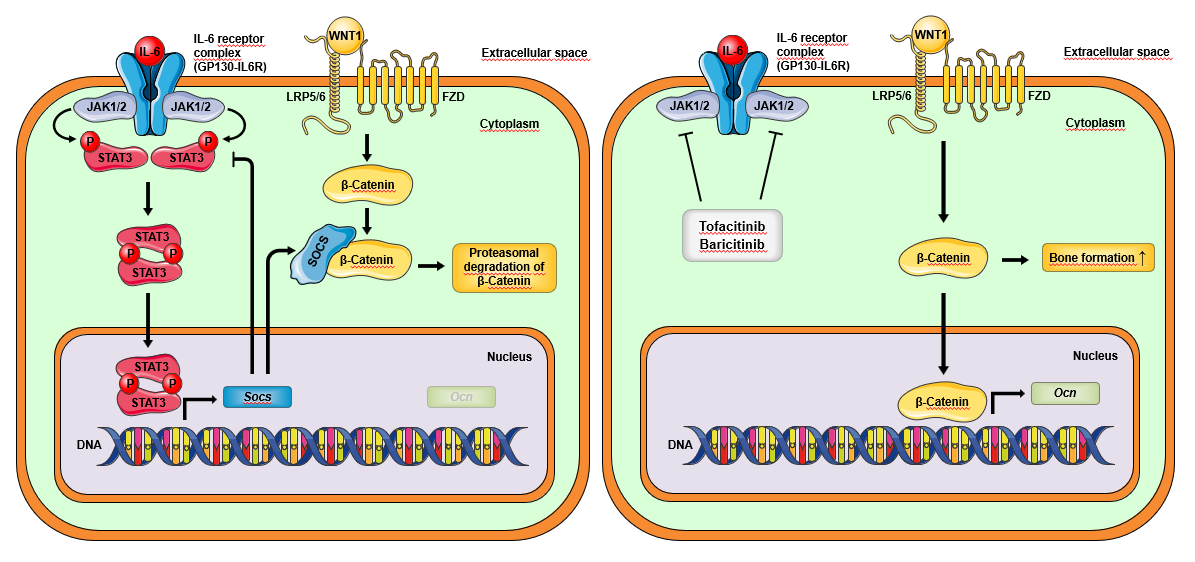
Systemic Effects of JAK Inhibitors on Bone
It has been demonstrated that the osteoblast- and osteocyte-specific knockdown of STAT3 reduced bone mass and quality [28,29]. Therefore, we did not expect to find STAT3 downregulation being involved in JAK inhibitor-mediated effects on osteoblasts. However, one has to keep in mind, that even a cell-specific knockout of a universal signal transducer, such as STAT3, may exhibit different effects compared to the targeted downregulation of an upstream JAK protein. In the case of JAK inhibition STAT3 is still present and able to engage in signal transduction, as long as the inducing cytokine utilizes a JAK protein, not targeted by the drug. Since STAT3 is a prominent mediator of cytokine signaling of the IL-6 family of cytokines, which have been described to dampen osteoblast activity, we suspect that JAK inhibitors enhance osteoblast mineralization by interfering with IL-6 signaling [44]. Reduction in IL-6 signaling diminishes STAT3- mediated transcription of target genes, such as Rankl and Socs, which would otherwise engage mechanisms to counter bone formation [42,45].
Moreover, the effects of JAK inhibition need to be seen in a systemic context: While the in vitro investigations of osteoblast cultures rely solely on their intrinsic secretion machinery, in vivo osteoblasts are embedded in a framework of other cells and extracellular matrix which is laced with cytokines. We demonstrated a robust increase in bone mass upon JAK inhibition in a variety of preclinical models. To what extent our in vitro analysis of osteoclast and osteoblast monocultures is congruent with the systemic in vivo effects of JAK inhibitors remains to be determined. In vivo, osteoclasts could also be affected by JAK inhibition. Investigations have shown that osteoclasts are negatively affected by JAK inhibition indirectly via osteoblasts [45]. The group of Nakamura investigated osteoblast-osteoclast co-cultures and described that baricitinib reduced the expression of the receptor activator of nuclear factor-κB ligand (Rankl) in osteoblasts. With RANKL being one of the key drivers of osteoclast differentiation, osteoclastogenesis was impaired by baricitinib in osteoblast-osteoclast co-cultures [45]. Moreover, how osteocytes, an abundant cellular agonist of bone metabolism, can be affected with respect to JAK inhibition has not been well investigated (Figure 2).
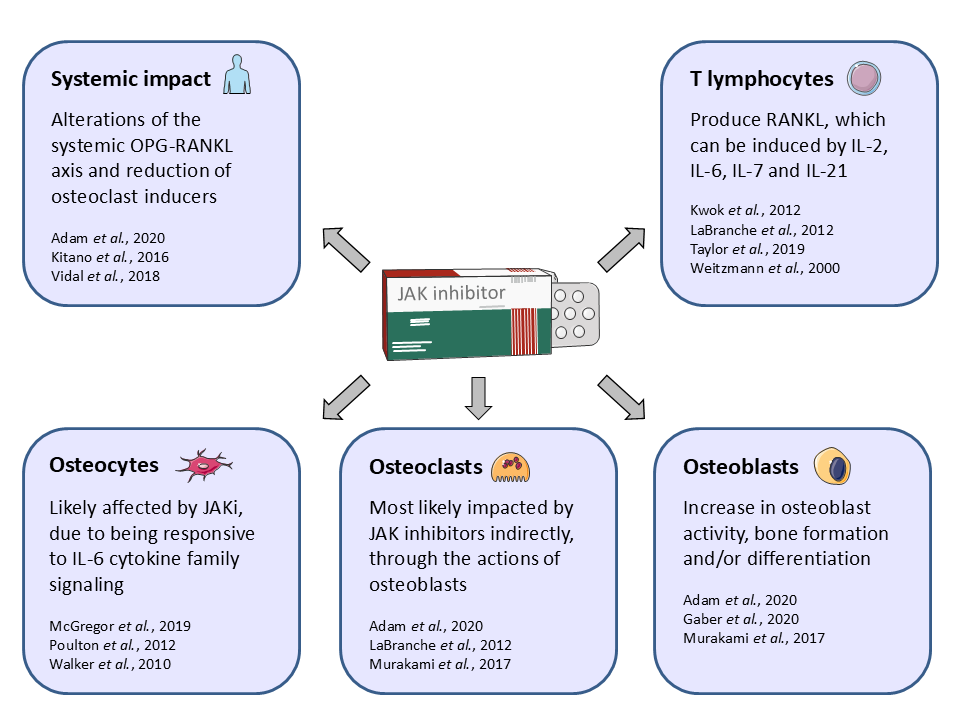
Summary
The fact that JAK inhibitors have osteoanabolic properties raises several possibilities, which need to be addressed in future research. For instance, the data raises the question whether JAK inhibitors augment systemic bone mass in humans with postmenopausal osteoporosis or inflammation-induced osteoporosis. As JAK inhibition targets multiple cytokines simultaneously, they may be able to restore a homeostatic bone environment. The observation that JAK inhibitors allow the repair of bone erosions in RA supports the notion that the osteoanabolic effect of JAK inhibitors is also relevant to patients.
Funding
This entirety of this research project was enabled by research grants from Eli Lilly and Pfizer, the Deutsche Forschungsgemeinschaft (projects DFG-CRC1181- project A05, SCHE (1583/14-1), FOR 2886 PANDORA, HA 8163/1-1), the Bundesministerium für Bildung und Forschung (METARTHROS), the European Union (ERC StG 640087–SOS, ERC 4D NanoScope) and the EU/ EFPIA Innovative Medicines Initiative 2 Joint Undertaking RTCure grant 777357, and the Interdisciplinary Center for Clinical Research at the Universitätsklinikum Erlangen of the FAU.
Conflicts of Interest Declaration
Authors declare no conflicts of interest.
References
2. Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. 2002 May 31;296(5573):1653-5.
3. Hammarén HM, Virtanen AT, Raivola J, Silvennoinen O. The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine. 2019 Jun 1;118:48-63.
4. Amaya-Amaya, J., Rojas-Villarraga, A., Mantilla, R.D., and Anaya, J.-M. (2013). Chapter 24: Rheumatoid arthritis. In Autoimmunity: From Bench to Bedside, J.M.Anaya, Shoenfeld, Y., Rojas-Villarraga, A., Levy, R. A.,Cervera, R., ed. (Bogota (Colombia): El Rosario University Press © 2013 Universidad del Rosario.).
5. Lee DM, Weinblatt ME. Rheumatoid arthritis. Lancet.2001; 358, 903-911.
6. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK–STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. 2017 Apr 1;77(5):521-46.
7. Hodge JA, Kawabata TT, Krishnaswami S, Clark JD, Telliez JB, Dowty ME, et al. The mechanism of action of tofacitinib-an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis. Clinical and Experimental Rheumatology. 2016 Mar 1;34(2):318-28.
8. EMA (2016). European public assessment reports: Olumiant (Baricitinib) (European Medicines Agency - Science, Medicines, Health).
9. EMA (2017). European public assessment reports: Xeljanz (Tofacitinib) (European Medicines Agency - Science, Medicines, Health).
10. EMA (2019). European public assessment reports: Rinvoq (Upadacitinib) (European Medicines Agency - Science, Medicines, Health).
11. EMA (2020). European public assessment reports: Jyseleca (Filgotinib) (European Medicines Agency - Science, Medicines, Health).
12. FDA (2012). Drugs@FDA: FDA-Approved Drugs: Xeljanz (Tofacitinib citrate) (United States Food and Drug Administration).
13. MFDA (2018). Drugs@FDA: FDA-Approved Drugs: Olumiant (Baricitinib) (United States Food and Drug Administration).
14. FDA (2019). Drugs@FDA: FDA-Approved Drugs: Rinvoq (Upadacitinib) (United States Food and Drug Administration).
15. Changelian PS, Flanagan ME, Ball DJ, Kent CR, Magnuson KS, Martin WH, et al. Prevention of organ allograft rejection by a specific Janus kinase 3 inhibitor. Science. 2003 Oct 31;302(5646):875-8.
16. Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus Kinase (JAK) inhibitors for inflammatory diseases: Miniperspective. Journal of Medicinal Chemistry. 2014 Jun 26;57(12):5023-38.
17. McInnes IB, Byers NL, Higgs RE, Lee J, Macias WL, Na S, et al. Comparison of baricitinib, upadacitinib, and tofacitinib mediated regulation of cytokine signaling in human leukocyte subpopulations. Arthritis Research & Therapy. 2019 Dec;21(1):1.
18. Parmentier JM, Voss J, Graff C, Schwartz A, Argiriadi M, Friedman M, et al. In vitro and in vivo characterization of the JAK1 selectivity of upadacitinib (ABT-494). BMC Rheumatology. 2018 Dec;2(1):1-1.
19. Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. The Journal of Immunology. 2013 Oct 1;191(7):3568-77.
20. Lee YH, Song GG. Relative efficacy and safety of tofacitinib, baricitinib, upadacitinib, and filgotinib in comparison to adalimumab in patients with active rheumatoid arthritis. Zeitschrift für Rheumatologie. 2020 Feb 13:1.
21. Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nature Reviews Drug Discovery. 2017 Dec;16(12):843.
22. Taylor PC, Keystone EC, van der Heijde D, Weinblatt ME, del Carmen Morales L, Reyes Gonzaga J, et al. Baricitinib versus placebo or adalimumab in rheumatoid arthritis. New England Journal of Medicine. 2017 Feb 16;376(7):652-62.
23. Li J. JAK-STAT and bone metabolism. Jak-stat. 2013 Jul 15;2(3):e23930.
24. Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996 Feb 9;84(3):443-50.
25. Kim S, Koga T, Isobe M, Kern BE, Yokochi T, Chin YE, et al. Stat1 functions as a cytoplasmic attenuator of Runx2 in the transcriptional program of osteoblast differentiation. Genes & Development. 2003 Aug 15;17(16):1979-91.
26. Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, et al. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK–STAT signaling pathway. Cell. 1996 Feb 9;84(3):431- 42.
27. Takeda K, Noguchi K, Shi W, Tanaka T, Matsumoto M, Yoshida N, et al. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proceedings of the National Academy of Sciences. 1997 Apr 15;94(8):3801-4.
28. Itoh S, Udagawa N, Takahashi N, Yoshitake F, Narita H, Ebisu S, et al. A critical role for interleukin-6 familymediated Stat3 activation in osteoblast differentiation and bone formation. Bone. 2006 Sep 1;39(3):505-12.
29. Zhou H, Newnum AB, Martin JR, Li P, Nelson MT, Moh A, et al. Osteoblast/osteocyte-specific inactivation of Stat3 decreases load-driven bone formation and accumulates reactive oxygen species. Bone. 2011 Sep 1;49(3):404-11.
30. Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, et al. Hyper-IgE syndrome with recurrent infections—an autosomal dominant multisystem disorder. New England Journal of Medicine. 1999 Mar 4;340(9):692-702.
31. He J, Shi J, Xu X, Zhang W, Wang Y, Chen X, et al. STAT3 mutations correlated with hyper-IgE syndrome lead to blockage of IL-6/STAT3 signalling pathway. Journal of Biosciences. 2012 Jun 1;37(2):243-57.
32. Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, et al. STAT3 mutations in the hyper-IgE syndrome. New England Journal of Medicine. 2007 Oct 18;357(16):1608-19.
33. Rael EL, Marshall RT, McClain JJ. The hyper-IgE syndromes: lessons in nature, from bench to bedside. World Allergy Organization Journal. 2012 Dec;5(7):79-87.
34. Hans D, Ochs CIES, Jennifer M. Puck (2006). Primary Immunodeficiency Diseases: A Molecular & Cellular, 2nd edition (Oxford University Press).
35. Maruotti N, Corrado A, Rotondo C, Cantatore FP. Janus kinase inhibitors role in bone remodeling. Journal of Cellular Physiology. 2020 Mar;235(3):1915-20.
36. Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, et al. Dominant-negative mutations in the DNAbinding domain of STAT3 cause hyper-IgE syndrome. Nature. 2007 Aug;448(7157):1058-62.
37. Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak–STAT signaling in the immune system. Nature Immunology. 2017 Apr;18(4):374.
38. Cohen SB, Dore RK, Lane NE, Ory PA, Peterfy CG, Sharp JT, et al. Denosumab treatment effects on structural damage, bone mineral density, and bone turnover in rheumatoid arthritis: a twelve-month, multicenter, randomized, double-blind, placebo-controlled, phase II clinical trial. Arthritis & Rheumatism. 2008 May;58(5):1299-309.
39. Schett G, Gravallese E. Bone erosion in rheumatoid arthritis: mechanisms, diagnosis and treatment. Nature Reviews Rheumatology. 2012 Nov;8(11):656.
40. Zerbini CA, Clark P, Mendez-Sanchez L, Pereira RM, Messina OD, Uña CR, et al. Biologic therapies and bone loss in rheumatoid arthritis. Osteoporosis International. 2017 Feb;28(2):429-46.
41. Orsolini G, Bertoldi I, Rossini M. Osteoimmunology in rheumatoid and psoriatic arthritis: potential effects of tofacitinib on bone involvement. Clinical Rheumatology. 2020 Mar;39(3):727-36.
42. Adam S, Simon N, Steffen U, Andes FT, Scholtysek C, Müller DI, et al. JAK inhibition increases bone mass in steady-state conditions and ameliorates pathological bone loss by stimulating osteoblast function. Science Translational Medicine. 2020 Feb 12;12(530).
43. Les Laboratoires Servier, S. (2020). SMART - Servier Medical Art. Available at: https://smart.servier.com/, accessed: 19. January 2021.
44. Kaneshiro S, Ebina K, Shi K, Higuchi C, Hirao M, Okamoto M, et al. IL-6 negatively regulates osteoblast differentiation through the SHP2/MEK2 and SHP2/ Akt2 pathways in vitro. Journal of Bone and Mineral Metabolism. 2014 Jul 1;32(4):378-92.
45. Murakami K, Kobayashi Y, Uehara S, Suzuki T, Koide M, Yamashita T, et al. A Jak1/2 inhibitor, baricitinib, inhibits osteoclastogenesis by suppressing RANKL expression in osteoblasts in vitro. PLoS One. 2017 Jul 14;12(7):e0181126.
46. Gaber T, Brinkman AC, Pienczikowski J, Diesing K, Damerau A, Pfeiffenberger M, et al. Impact of Janus Kinase Inhibition with Tofacitinib on Fundamental Processes of Bone Healing. International Journal of Molecular Sciences. 2020 Jan;21(3):865.
47. Kitano M, Kitano S, Sekiguchi M, Azuma N, Hashimoto N, Tsunoda S, et al. AB0394 early effect of tofacitinib on osteoclast regulator in rheumatoid arthritis. 2016;75:1040.
48. Kwok SK, Cho ML, Park MK, Oh HJ, Park JS, Her YM, Lee SY, Youn J, Ju JH, Park KS, Kim SI. Interleukin-21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen-induced arthritis. Arthritis & Rheumatism. 2012 Mar;64(3):740-51.
49. LaBranche TP, Jesson MI, Radi ZA, Storer CE, Guzova JA, Bonar SL, et al. JAK inhibition with tofacitinib suppresses arthritic joint structural damage through decreased RANKL production. Arthritis & Rheumatism. 2012 Nov;64(11):3531-42.
50. McGregor NE, Murat M, Elango J, Poulton IJ, Walker EC, Crimeen-Irwin B, et al. IL-6 exhibits both cis-and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. Journal of Biological Chemistry. 2019 May 10;294(19):7850-63.
51. Poulton IJ, McGregor NE, Pompolo S, Walker EC, Sims NA. Contrasting roles of leukemia inhibitory factor in murine bone development and remodeling involve regionspecific changes in vascularization. Journal of Bone and Mineral Research. 2012 Mar;27(3):586-95.
52. Taylor PC. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology. 2019 Feb 1;58(Supplement_1):i17-26.
53. Vidal B, Cascão R, Finnilä MA, Lopes IP, da Glória VG, Saarakkala S, Zioupos P, Canhão H, Fonseca JE. Effects of tofacitinib in early arthritis-induced bone loss in an adjuvant-induced arthritis rat model. Rheumatology. 2018 Aug 1;57(8):1461-71.
54. Walker EC, McGregor NE, Poulton IJ, Solano M, Pompolo S, Fernandes TJ, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. The Journal of Clinical Investigation. 2010 Feb 1;120(2):582-92.
55. Weitzmann MN, Cenci S, Rifas L, Brown C, Pacifici R. Interleukin-7 stimulates osteoclast formation by upregulating the T-cell production of soluble osteoclastogenic cytokines. Blood, The Journal of the American Society of Hematology. 2000 Sep 1;96(5):1873-8.