Abstract
Understanding the development of the auditory system is crucial for uncovering the molecular origins of hearing and its related disorders. During this development, spiral ganglion neurons extend peripheral fibers to cochlear hair cells and central projections to the cochlear nuclei, setting up a tonotopic map that connects the ear to the brainstem, enabling frequency-specific sound perception. This sensory information is then integrated bilaterally through a relay involving the superior olivary complex, lateral lemniscus, inferior colliculus, medial geniculate body, and the auditory cortex. While anatomical connectivity has been well-documented, recent advancements have revealed gene regulatory networks that coordinate the specification, differentiation, and connectivity of auditory neurons. In this review, we examine the molecular cascades guiding auditory system development, emphasizing transcriptional hierarchies and lineage determinants. Insights into these mechanisms enhance our understanding of auditory circuit formation and provide a critical foundation for therapeutic strategies aimed at addressing congenital and acquired hearing loss.
Keywords
Spiral ganglion neurons, Cochlear nuclei, Cochlear hair cells, Brainstem, Genetic basis, Auditory cortex
Introduction
Mammals are the only vertebrates known for their ability to hear high frequencies above 10 kHz. They possess a unique organization of cochlear hair cells (HCs), which are divided into inner and outer hair cells (IHCs and OHCs) that receive innervation from spiral ganglion neurons (SGNs). SGNs transmit auditory signals to the cochlear nuclei (CNs), which relay the information to higher auditory processing centers, including the auditory forebrain. This overview addresses the genetic mechanisms that establish tonotopic organization in the auditory system: IHCs connect the brain to SGNs, which are organized in the cochlea in a frequency-specific manner, with high frequencies at the base and low frequencies at the apex. SGNs project centrally to CN neurons, which are also tonotopically organized, preserving the frequency map from the periphery to the brainstem. Reciprocal connections ease the encoding, transmission, and interpretation of auditory information. Recent studies have enhanced our understanding of the genetic basis for the development of the auditory system and the molecular processes that shape hearing function, paving the way for novel therapeutic strategies that may alleviate hearing loss in children and older adults [1,2]. Recent gene therapy clinical trials suggest that targeting specific genetic mutations, such as TMC2 and OTOF, could enable the restoration of hearing in affected individuals [3,4].
The review will outline the genetic basis for the cochlea, brainstem, midbrain, and cortex, which define the molecular basis for hearing. We try to integrate specific mechanistic overlaps that present more recent approaches to guide future research detailing the effect of central projections without tonotopic organization. We provide literature citations of recent papers that may inform future research directions in cochlear and central nervous system development and guide therapeutic approaches.
Building Spiral Ganglion Neurons to Connect the Cochlear Nuclei with Hair Cells
Molecular and cellular processes govern the development, differentiation, and maintenance of cochlear hair cells (HCs), spiral ganglion neurons (SGNs), and cochlear nuclei (CNs). Understanding the recurring roles of key transcription factors requires an integrated approach, including in situ hybridization and immunohistochemistry, as well as gene knockout mouse models, to reveal their spatiotemporal regulation during auditory pathway development. HCs, SGNs, and CNs differentiate as distinct but temporally overlapping hubs. SGNs proliferate and develop in a base-to-apex gradient from embryonic day E10.5 to E12.5. In contrast, while HC cell cycle progression follows the opposite direction, from the apex (E12.5) to the base (E13.5), HC differentiation initiates near the cochlear base around E13.5 and continues toward the apex until E18.5 [5]. CN neurons proliferate between approximately E10 and E14, overlapping with SGNs and HCs. SGNs and HCs are derived from the ear placode, while CN neurons are derived from rhombomeres (r2-r5) from the dorsal brainstem [6]. SGN fibers reach the CN at E12 at the basal turn, whereas the topology between the basal and apical turn afferents is established at E14.5 (Figure 1). SGN fibers reach IHCs via type I fibers starting at E15 and then OHCs via type II fibers at E18 [7]. While SGNs send input to the CN, the fibers project to several subnuclei, the anteroventral cochlear nucleus (AVCN), posteroventral cochlear nucleus (PVCN), and dorsal cochlear nucleus (DCN; Figure 1), each with distinct anatomical and functional characteristics important for frequency tuning, intensity coding, and temporal processing [8].
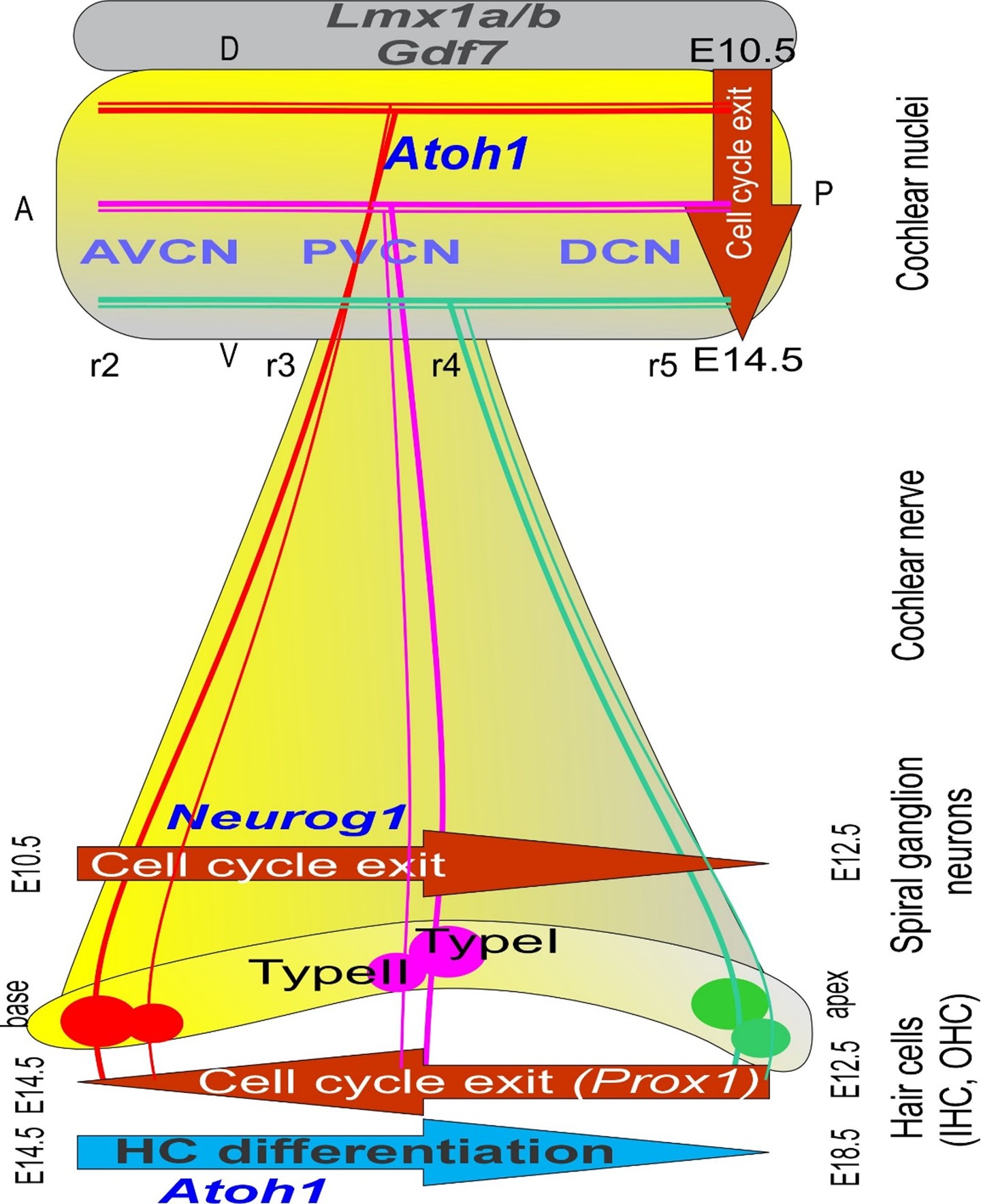
Figure 1. The proliferation of hair cells, spiral ganglion neurons, and cochlear nuclei is displayed. Neurog1 is expressed first and progresses from the base to the apex. Proliferation overlaps with cochlear nuclei from dorsal to more ventral regions. The proliferation of hair cells begins in the apex (Prox1) but upregulates later (Atoh1) to reach the apex much later. The first connection originates from the basal turn to innervate the dorsal part of the cochlear nuclei, showing a delay of inner hair cell development, while the apex extends to the ventral cochlear nuclei, demonstrating a significantly later progression of apical hair cell formation. Two types of fibers connect the hair cells and the cochlear nuclei: the thinner type II fibers. AVCN: Anteroventral Cochlear Nuclei; Atoh1: Atonal homolog 1; DCN: Dorsal Cochlear Nuclei; Gdf7: Growth differentiation factor 7; Neurog1: Neurogenin 1; Lmx1a/b: LIM homeobox transcription factor 1 alpha/1 beta; Prox1: Prospero homeobox 1; PVCN: Posteroventral Cochlear Nuclei. Modified from [68].
SGNs depend on Eya1/Smarca4, Sox2, and Neurog1
Upstream, the HDACs are responsible for neuronal differentiation, shedding light on HDAC and REST inhibitors as potential targets for use as therapeutic agents for the treatment of neurodegenerative diseases [9,10]. Eya1/Six1/Smarca4 and Sox2 define the neurosensory domain of the inner ear precursors, whereas Neurog1 initiates neuronal development [11,12]. A unique trio of transcription factors, Tbx1-3, in concert with Neurog1, regulates otocyst neurogenesis [13]. The transcriptional repressor Tbx1 acts as a selective gene, controlling neuronal fate in the otocyst, while deletions of both Tbx2 and Tbx3 expand the otic neurogenic domain and disrupt inner ear morphogenesis. Conditional deletions of Gata3, Lmx1a/b, Dicer, or Shh result in a complete loss of SGNs [14-16], while nearby developing vestibular neurons form. Deletions of the transcription factors Neurod1 and Isl1 result in abnormal migration of SGNs [17-19]. Neurotrophins are needed for SGN viability and maturation [20], with the loss of all SGNs occurring in the absence of the neurotrophins encoded by Bdnf and Ntf3 or the genes encoding their receptors, TrkB and TrkC [21]. A possible downstream effect provides evidence for a pathophysiological mechanism of deafness and shows how genes involved in different forms of deafness may interact together, including Bdnf/Ntf3 [22,23].
Type I SGNs can be classified into three major subtypes: type Ia-c [24]. Type Ia SGNs are characterized by enriched expression of Calb2 and Pcdh20, which later becomes Runx1-positive [25] . Type Ib SGNs show enriched expression of Calb1, while type Ic SGNs show expression of Pou4f1. Type Ia afferents project preferentially to the pillar aspects of the IHCs [24,26] and correspond to a high spontaneous rate, whereas type Ic SGNs project preferentially to the modiolar face of the IHCs and correspond to low spontaneous rate fibers. Type Ia, Ib, and Ic SGNs show subtle variations in their distribution along the apical, middle, and basal turns of the cochlea. Approximately 7% of SGN afferents form type II neurons that are selectively Prph-positive at a specific developmental stage. The type II neurons innervate OHCs and also reach out to the CN [7]. All hair cells depend on Otoferlin (Otof), which is needed for glutamate release. Without Otoferlin, the proper activation of the SGNs or CNs is profoundly disrupted [27,28]. Indeed, human mutations of OTOF can be rescued by inserting the mutant gene with the normal OTOF form and restoring hearing [3].
HCs depend on Eya1/Smarca4, Sox2, and Atoh1
Like SGNs, HC progenitors initiate development through the activity of transcriptional regulators Eya1, Six1, Hdac’s, Rest and Smarca [10,11]. Downstream expression of the transcription factor Sox2 is required to initiate HC formation [12]. Atoh1 is obligatory for further differentiation of HCs [29]. Downstream genes include the transcriptional regulators (Pou4f3, Gfi1, and Barhl1), which are necessary for HC maintenance [30]. Deletions of the transcription factors Gata3, Lmx1a/b, or Pax2 result in the absence of all HCs [14,15,31]. Loss of the bHLH transcription factor Neurog1, and other regulatory factors such as Lmx1a or Foxg1, disrupts Atoh1 expression and impairs HC differentiation, resulting in a shortened cochlea with multiple rows of HCs and supporting cells, or a fate switch from cochlear to vestibular-like HCs, as shown in Foxg1, Neurog1, and Lmx1a mutants [29,32,33]. Irx3/5 DKO mice show a fusion of the basal cochlear turn with the saccule, the development of vestibular-like HCs with an incomplete segregation of topological projections to the cochlear nuclei [34]. A shorter cochlea and conversions of OHCs into IHCs are associated with the deletion of Neurod1 and Isl1 [18,35]. Tbr2 (aka Eomes) regulates the cycling of neurons and hair cells by interacting with Dll, Notch and Hes [36,37]. The differentiation between IHCs and OHCs depends on distinct genetic programs. OHC development requires downstream expression of the transcriptional repressor Insm1 and transcriptional regulator Ikzf2 [38]. Loss of the transcriptional repressor Tbx2 converts IHC into OHC-like HCs; a similar transformation is documented with Bcl11a deletion [39]. In contrast, IHCs do not develop without Srrm3/4 [40]. Many more null mutations are presented in recent papers that detail the null deletion of Meis2. Results suggest that several additional gene expressions may be required to develop new hair cells [41,42].
CN neurons depend on Atoh1 and Ptf1a
While SGNs and HCs show limited variability, CN neuron populations arise from distinct progenitor domains, notably Atoh1 and Ptf1a, that generate dA1 (Atoh1) and dA4/dB1 (Ptf1a) neurons. Spherical bushy cells form a large calyx in r2, while globular bushy cells interact with significant gaps of the calyx and belong r3 [43,44]. D-, L-, and T-stellate cells likely derive from r3 [8], with T-stellate cells being glutamatergic and D-stellate cells glycinergic. The r4 neuronal populations are represented by octopus cells (PVCN) and giant cells, which are dependent on Atoh1 expression [45]. The r5 populations include Atoh1-positive-derived neurons (unipolar brush cells, fusiform cells, and granule cells) and a large added subpopulation of Ptf1a+-positive-derived neurons (GABAergic Golgi cells, superficial stellate cells, glycinergic cartwheel, and tuberculoventral cells). Type I SGNs connect with Atoh1-positive-derived neurons in the CN as end bulbs or end in small connections within the topological organization. The type I and type II fibers are parallel, but type II fibers only reach the granule cells [46]. All glutamate neurons in the CN derive from the Atoh1-positive domain (spherical, globular, T-stellate, roof, octopus, fusiform, giant, unipolar brush cells (UBCs), and granule cells). Neurons that are derived from the Ptf1a-positive domain are either glycinergic (possibly from dA4; D-stellate, cartwheel, and tuberculoventral cells) or GABAergic (possibly from dB1; Golgi, superficial stellate cells [6]). Moreover, Otof is required for the proper activation of cochlear hair cells and cochlear nuclei to allow functional interaction [47].
Loss of Ptf1a results in the overproduction of neurons from the Lmx1b lineage (Figure 1), rather than Lhx1/5 and Pax2 expression in dB1 neurons [6,48]. Conversion shows a graded effect that turns r2-3 into Lmx1b-positive neurons instead of dB1, while r4-6 transforms from dA4 into dA3 in the Ptf1a-/-. Utilizing either Egr2-cre or Hoxb1-cre [40] corresponds with the origin of dA1 and dA4/dB1-derived neurons [48]. Atoh1 deletion causes the loss of r3- and r5-originated neurons and nearly complete elimination of the AVCN in Egr2-cre; Atoh1CKO (conditional knockout) (Figure 2), while most r4 neurons from the PVCN and DCN are lost in Hoxb1-cre; Atoh1CKO. Deletion of Bhlhb5 removes two distinct types of DCN neurons: unipolar brush cells and cartwheel cells. Furthermore, Lbx1 is expressed in stellate and cartwheel cells of the DCN, but not in Golgi cells, implying a diversification of genetic programs among distinct types of inhibitory neurons, specifically Ptf1a-lineage neurons [6]. More recent analysis has found distinct neuronal populations with unique gene expression [8] that require further investigation to connect their origin (Atoh1; Ptf1a) with their adult expression. The absence of Neurod1 or Isl1 results in disorganized SGN fibers that connect with reduced input, allowing for an unusual tonotopic organization [18,19]. The lack of Ptf1a likely results in the absence of inhibitory GABAergic neurons, which can disrupt normal function by disrupting Cl- homeostasis [49].
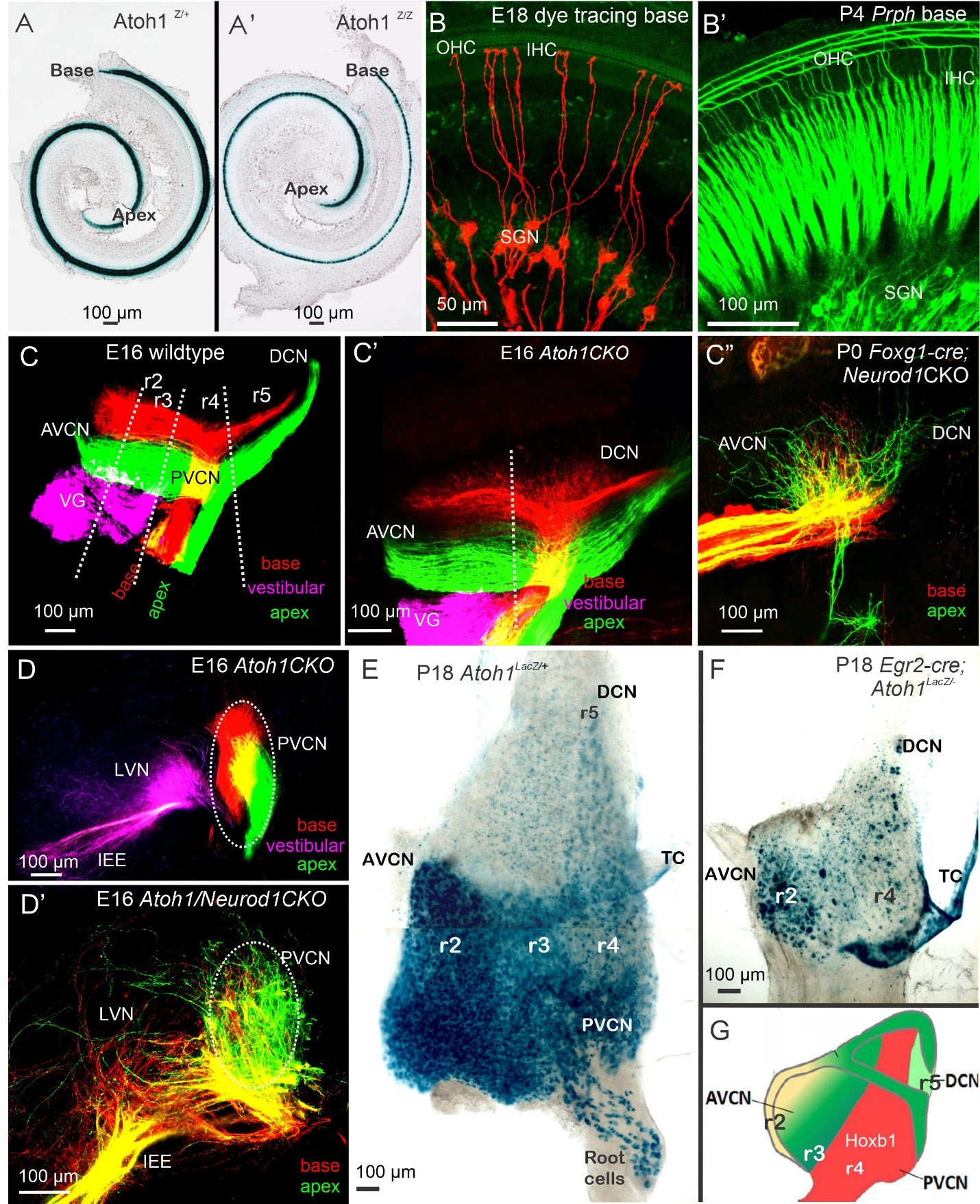
Figure 2. Development of SGNs, HCs, and central projection. All hair cells depend on Atoh1 (A), which initially develop even without Atoh1 expression (A’). Applying dye tracing selectively labels the SGNs that innervate the IHC (B). Prph expression indicates the innervation of OHCs from type II fibers (B’). The central projection displays the tonotopic organization in control mice (C). Even in the presence of Atoh1, fibers can develop to innervate the cochlear nuclei (C’). Neurod1 deletions show overlapping fibers that innervate the small CN (C”). Sections reveal segregation in Atoh1 null mice (D), while the absence of Atoh1 and Neurod1 results in overlap (D’). A flat mount of the CN using Atoh1LacZ indicates the four rhombomeres with a gradient of Atoh1 (E). Combining Atoh1-Lacz with genetic engineering using Egr2-cre (also known as Krox20) demonstrates the absence of r3 and r5, and a reduced generation of r2 and r4 (F). In red, the Hoxb1-positive neurons generate r4 neurons. Abbreviations: Atoh1: Atonal homolog 1; AVCN: Anteroventral Cochlear Nuclei; DCN: Dorsal Cochlear Nuclei; Egr2: Early growth response 2; HCs: Hair Cells; IHC: Inner Hair Cells; Neurod1: Neurogenic differentiation 1; OHC: Outer Hair Cells; Prph: Peripherin; PVCN: Posteroventral Cochlear Nuclei; r2-r5: Rhombomere 2- rhombomere 5; SGNs: Spiral Ganglion Neurons; TC: Tela Choroidea; VG: Vestibular Ganglion Neurons. Taken from [2,35,44,50,69].
Overall, SGNs rely on Neurog1, while cochlear HCs and CN neurons depend on Atoh1, with Ptf1a also being involved in the CN (Figure 3). The cochlear system evolves from a relatively simple gene expression program into a specialized structure, consisting of only four neuronal subtypes that innervate just two types of HCs. This contrasts with the greater neuronal diversity of the CN, which includes at least 12 distinct neuron types. Each input is segregated and overlaps with distinct projections that coordinate functional expression across various gene families, thereby customizing projection patterns. Note that the central projection is tonotopic, receiving fibers from the base, middle, and apex to provide frequency-specific input.
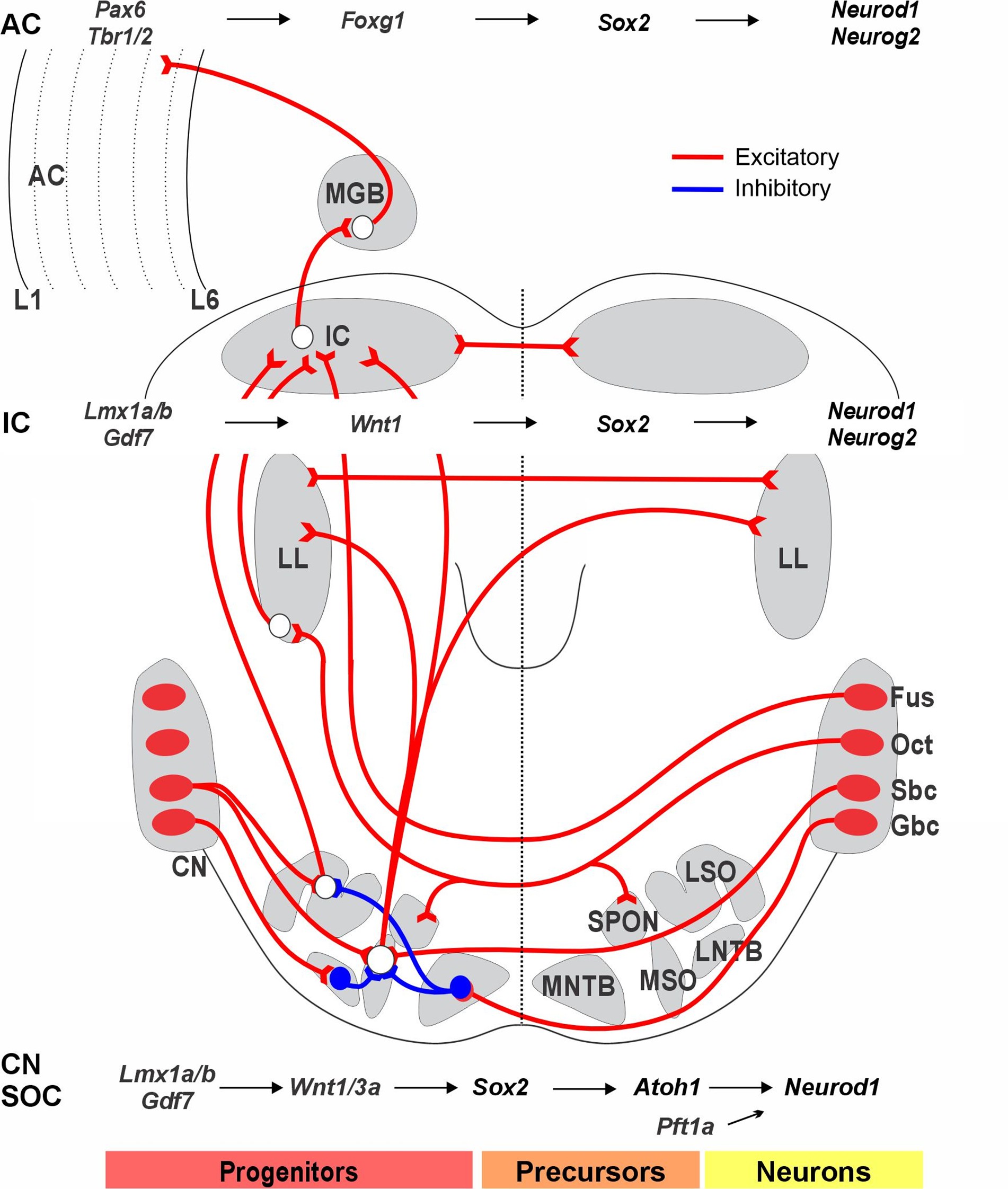
Figure 3. The output of neurons in the cochlear nucleus (CN) projects to the superior olivary complex (SOC). CN/SOC depend on Lmx1a/b, Gdf7, and Wnt1/3a, which guide the migration of SOC from r4. Bilateral interactions involve the lateral superior olive (LSO), medial superior olive (MSO), medial nucleus of the trapezoid body (MNTB), and lateral nucleus of the trapezoid body (LNTB): LSO receives binaural input from the ipsilateral spherical bushy cells (SBCs) and contralateral globular bushy cells (GBCs), which is inhibited by neurons from the MNTB. MSO receives bilateral excitatory input from both ipsilateral and contralateral SBCs and inhibitory input from GBCs via the MNTB and LNTB. The octopus cells (OCT) provide bilateral input to the superior paraolivary nucleus (SPON) and connect with the lateral lemniscus (LL). The dorsal cochlear nucleus, originating from fusiform neurons, bypasses the SOC to reach the contralateral inferior colliculus (IC). Progenitors in the IC depend on Lmx1a/b, Gf7, Wnt1, and Sox2 to generate neurons, which then rely on Neurod1 and Neurog2 to differentiate into excitatory glutamatergic neurons. Additionally, the medial geniculate body (MGB) receives input from the IC and projects to the auditory cortex (AC). Progenitors in the auditory cortex depend on Pax6, Tbr1/2, and Foxg1 to induce Sox2 to create neurons that depend on Neurod1 and Neurog2 to differentiate into excitatory glutamatergic neurons. These interactions play significant roles in processing auditory information along the neural pathway. The molecular factors guiding the development of these projection pathways are poorly understood. Images are compiled from various sources. Modified after [2].
Transformation of Unilateral Sound Input into Bilateral Auditory Information through SOC, IC, MGB, and AC Interactions
The superior olivary complex (SOC) receives auditory information from the CN and serves as the foundation for binaural auditory processing (Figure 3). Neurons destined for the SOC are generated between embryonic day 10.5 (E10.5) and embryonic day 17.5 (E17.5). During this time, they primarily migrate, differentiate, and organize to form the SOC [50]. The SOC neurons are partially dependent on Atoh1 and the homeobox-containing gene En1 [51], with a sizable number originating in r4 and 5. An incomplete deletion of rhombomeres 3/5 using Krox20 (Egr2)-cre deletion of Atoh1 reveals a small remaining population with reduced SOC input [44]. It stays unclear how many SOC neurons are lost directly in the absence of the CN.
The inferior colliculus (IC) serves as a key integration center for auditory input, receiving signals from both the CN and SOC and directing its output to the medial geniculate body (MGB). Various genes play crucial roles in the development of the IC [52]. The gene Opb encodes a small GTPase of the Ras superfamily that antagonizes Shh, a vital signaling molecule involved in the early patterning of the IC [52]. A complete loss of Wnt1 acts downstream of Lmx1b, leading to the elimination of the entire midbrain (Figure 3). Deletions of Fgf8 and Pax2, among others, reduce the volume of the IC [53,54]. In contrast, overexpression of Shh through Pax2-cre-driven expression of Smo, which encodes a G protein-coupled receptor, results in exaggerated development of both the IC and the cerebellum [55]. The expression of Isl1 under Pax2 regulatory sequences causes a modest reduction in the size of the IC and partially disrupts its normal function [56]. Downstream of Pax2 are Pax3, Pax7, and Meis2, which influence the development of the IC roof plate [57]. The expression of Otx2 is driven by Sox2, Ascl1, Neurod1, Neurog2, and Dll3, shaping the early initiation and differentiation of the IC [58]. The patterning gene Dbx1 is a crucial factor for the postnatal survival of the IC [54]. Beyond this early regulation, downstream glutamate, glycine, and GABAergic-expressing neurons in the IC require further investigation [19].
The diencephalon belongs to prosomere 2, and gene deletions reveal its dependence on Shh. Prosomere 2 is found caudally to the neuropore. It is separated from the zona limitans [59], which divides Foxg1 in prosomere 3 [60]. MGB development begins with connections from the IC forming between E13 and E18 before the onset of auditory sensory perceptions. Pax6 is crucial for the forebrain and the thalamus [61]. Early expressed genes include Wnt3, Tcf4, Meis2, and Irx3, some of which are essential for early development, such as Zic4 and Foxp2. Furthermore, Foxp2 is a key gene for speech and language development [62]. Tbr1 is expressed early and defines glutamatergic neurons in the forebrain. The connections of these neurons are partially affected by the targeted deletions of Pax6, Foxp2, Wnt3, Tcf4, and Irx3. Several downstream genes have been shown, including multiple bHLH genes (Neurog2, Ascl1, Olig2, and Neurod1). More analyses of conditional deletions are required to clarify the gene interactions within the thalamus [63].
The developing forebrain, including the auditory cortex (AC), is regulated by key transcription factors such as Otx1, Pax6, Shh, Emx2, and Foxg1 [61]. Additionally, a set of genes is necessary for the development of a normal forebrain, including Dlx, Nkx2.1, Tbx1, and Tbx2. Transcription factors vary in the developing forebrain in a precise, inside-out manner, establishing reproducible spatiotemporal patterns of gene regulatory networks (Figure 3). For example, Pax6 deletion alters the expression of genes such as Ascl1, Neurog2, Neurod1, Tbr1, Sox5, Sox9, and Hes5, while other genes like Foxg1 remain largely unaffected [61]. An opposing interaction between Pax6 and Foxg1, both of which depend on Shh and BMPs, regulates the competence of cortical cells. A double loss of Pax6 and Foxg1 abolishes Ascl1, Olig2, Gsx2, and Dlx1/2/3, which are redirected to a distinct neuronal variant. Neurog2 acts downstream to regulate Neurod1 expression, which is critical for the differentiation of hippocampal granule neurons [63].
In mice, AC neurons are generated between E11.5 and E13.5, with the earliest-born neurons being Cajal-Retzius neurons that develop slightly earlier, between E10.5 and E12.5. Neuronal migration plays a crucial role in the development of distinct cortical layers [64]. The early cortical structure, known as the preplate differentiates into two main regions: the marginal zone and the subplate zone [65]. Subplate neurons are the first to receive input from the thalamus, which is later redirected to cortical layer 4 in the adult brain. The maturation of inputs from cortical neurons shows a developmental delay in the AC, which undergoes functional reorganizations to form orientation columns. Initially, cortical layer 4 is innervated indirectly via the subplate, while direct thalamic input follows as development progresses. Delayed innervation of cortical layer 4 occurs after a first innervation of the subplate neurons. While most subplate neurons are lost in adults, a subset persists into adulthood within layer 6 [66]. Surviving subplate neurons may continue to influence altered or compensatory circuits. Disruptions in the precise timing, migration, or survival of AC neurons can cause neurological disorders [64]. Various activations primarily involve glutamatergic inputs to the subplate and layer 4, which receive both AMPA and NMDA receptor-mediated inputs, and local GABAergic neuronal interneurons also contribute to early inhibitory circuits. A progressive segregation is established in a discrete input/output relationship between the MGB and the AC. Further research is needed to fully map the reciprocal connections from the AC back to the MGB, midbrain, and brainstem, which likely contribute to feedback modulations and auditory plasticity [67].
Conclusions
This review provides an overview of the key genes required for the early development of the cochlea, brainstem, midbrain, and forebrain. Critical genes, such as Neurog1, which is essential for neuronal development, and Atoh1, which is required for hair cell and dorsal brainstem development, are indispensable. The deletion of these genes results not only in the absence of the cochlea and brainstem but may also affect the development of the midbrain and forebrain. Early cochlear fiber projections to the brainstem are crucial for establishing tonotopic organization, which refers to the spatial arrangement of sound frequency processing. Spiral ganglion neurons play a pivotal role in transmitting auditory signals from hair cells to the cochlear nuclei. However, bilateral projections from the cochlear nuclei form the basis for binaural hearing, allowing for the perception of sound localization and auditory integration that interacts with the cochlear nuclei, the superior olivary complex, the lateral lemniscus, and the inferior colliculi. This bilateral input continues to the forebrain and auditory cortex, which are crucial for complex auditory processing. We briefly present genetic mutations that disrupt these auditory pathways and discuss their potential implications for the development of auditory system processing. The forebrain, without input from the cochlear nuclei, requires a novel approach to selectively delete Neurog1 and/or Atoh1 that do not survive long enough to investigate the loss of this input on forebrain development.
Conflicts of Interest
None of the authors has a conflict of interest to disclose.
Funding Sources
ENY and BF were partly supported by National Institutes of Health grants AG060504 and AG051443. GP was partly supported by the Czech Science Foundation grant 23-05963S, and by the Czech Academy of Sciences RVO: 86652036.
Acknowledgment
We thank K. Elliott, J. Kersigo, and H.J. Lee for their valuable insights.
Author Contributions
All authors have contributed equally.
References
2. Pyott SJ, Pavlinkova G, Yamoah EN, Fritzsch B. Harmony in the Molecular Orchestra of Hearing: Developmental Mechanisms from the Ear to the Brain. Annu Rev Neurosci. 2024 Aug;47(1):1-20.
3. Brigande JV. Otoferlin gene therapy restores hearing in deaf children. Mol Ther. 2024 Apr 3;32(4):859-60.
4. Carlson RJ, Taiber S, Rubinstein JT. Gene Therapy for Hearing Loss: Which Genes Next? Otol Neurotol. 2025 Mar 1;46(3):239-47.
5. Fritzsch B, Dillard M, Lavado A, Harvey NL, Jahan I. Canal cristae growth and fiber extension to the outer hair cells of the mouse ear require Prox1 activity. PLoS One. 2010 Feb 23;5(2):e9377.
6. Elliott KL, Iskusnykh IY, Chizhikov VV, Fritzsch B. Ptf1a expression is necessary for correct targeting of spiral ganglion neurons within the cochlear nuclei. Neurosci Lett. 2023 May 29;806:137244.
7. Elliott KL, Kersigo J, Lee JH, Jahan I, Pavlinkova G, Fritzsch B, et al. Developmental Changes in Peripherin-eGFP Expression in Spiral Ganglion Neurons. Front Cell Neurosci. 2021 Jun 15;15:678113.
8. Jing J, Hu M, Ngodup T, Ma Q, Lau SN, Ljungberg MC, et al. Molecular logic for cellular specializations that initiate the auditory parallel processing pathways. Nat Commun. 2025 Jan 9;16(1):489.
9. Shukla S, Tekwani BL. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front Pharmacol. 2020 Apr 24;11:537.
10. Li J, Cheng C, Xu J, Zhang T, Tokat B, Dolios G, et al. The transcriptional coactivator Eya1 exerts transcriptional repressive activity by interacting with REST corepressors and REST-binding sequences to maintain nephron progenitor identity. Nucleic Acids Res. 2022 Oct 14;50(18):10343-10359.
11. Xu J, Li J, Zhang T, Jiang H, Ramakrishnan A, Fritzsch B, et al. Chromatin remodelers and lineage-specific factors interact to target enhancers to establish proneurosensory fate within otic ectoderm. Proc Natl Acad Sci U S A. 2021 Mar 23;118(12):e2025196118.
12. Dvorakova M, Macova I, Bohuslavova R, Anderova M, Fritzsch B, Pavlinkova G. Early ear neuronal development, but not olfactory or lens development, can proceed without SOX2. Dev Biol. 2020 Jan 1;457(1):43-56.
13. Kaiser M, Wojahn I, Rudat C, Lüdtke TH, Christoffels VM, Moon A, et al. Regulation of otocyst patterning by Tbx2 and Tbx3 is required for inner ear morphogenesis in the mouse. Development. 2021 Apr 15;148(8):dev195651.
14. Chizhikov VV, Iskusnykh IY, Fattakhov N, Fritzsch B. Lmx1a and Lmx1b are Redundantly Required for the Development of Multiple Components of the Mammalian Auditory System. Neuroscience. 2021 Jan 1;452:247-64.
15. Duncan JS, Fritzsch B. Continued expression of GATA3 is necessary for cochlear neurosensory development. PLoS One. 2013 Apr 16;8(4):e62046.
16. Riccomagno MM, Martinu L, Mulheisen M, Wu DK, Epstein DJ. Specification of the mammalian cochlea is dependent on Sonic hedgehog. Genes Dev. 2002 Sep 15;16(18):2365-78.
17. Mao Y, Reiprich S, Wegner M, Fritzsch B. Targeted deletion of Sox10 by Wnt1-cre defects neuronal migration and projection in the mouse inner ear. PLoS One. 2014 Apr 9;9(4):e94580.
18. Filova I, Bohuslavova R, Tavakoli M, Yamoah EN, Fritzsch B, Pavlinkova G. Early Deletion of Neurod1 Alters Neuronal Lineage Potential and Diminishes Neurogenesis in the Inner Ear. Front Cell Dev Biol. 2022 Feb 17;10:845461.
19. Filova I, Pysanenko K, Tavakoli M, Vochyanova S, Dvorakova M, Bohuslavova R, et al. ISL1 is necessary for auditory neuron development and contributes toward tonotopic organization. Proc Natl Acad Sci U S A. 2022 Sep 13;119(37):e2207433119.
20. Fritzsch B, Kersigo J, Yang T, Jahan I, Pan N. Neurotrophic factor function during ear development: expression changes define critical phases for neuronal viability. In: Dabdoub A, Fritzsch B, Popper A, Fay R, Editors. The primary auditory neurons of the mammalian cochlea. Springer Handbook of Auditory Research, vol 52. New York, NY: Springer International Publishing; 2016. P. 49-84.
21. Kersigo J, Fritzsch B. Inner ear hair cells deteriorate in mice engineered to have no or diminished innervation. Front Aging Neurosci. 2015 Mar 18;7:33.
22. Zhang H, Li H, Lu M, Wang S, Ma X, Wang F, et al. Repressor element 1-silencing transcription factor deficiency yields profound hearing loss through Kv7.4 channel upsurge in auditory neurons and hair cells. Elife. 2022 Sep 20;11:e76754.
23. Elliott KL, Kersigo J, Lee JH, Yamoah EN, Fritzsch B. Sustained Loss of Bdnf Affects Peripheral but Not Central Vestibular Targets. Front Neurol. 2021 Dec 16;12:768456.
24. Petitpré C, Faure L, Uhl P, Fontanet P, Filova I, Pavlinkova G, et al. Single-cell RNA-sequencing analysis of the developing mouse inner ear identifies molecular logic of auditory neuron diversification. Nat Commun. 2022 Jul 5;13(1):3878.
25. Shrestha BR, Wu L, Goodrich LV. Runx1 controls auditory sensory neuron diversity in mice. Dev Cell. 2023 Feb 27;58(4):306-19.e5.
26. Siebald C, Vincent PFY, Bottom RT, Sun S, Reijntjes DOJ, Manca M, et al. Molecular signatures define subtypes of auditory afferents with distinct peripheral projection patterns and physiological properties. Proc Natl Acad Sci U S A. 2023 Aug;120(31):e2217033120.
27. Moser T. Presynaptic physiology of cochlear inner hair cells. The Senses. 2020:441-67.
28. Chen H, Monga M, Fang Q, Slitin L, Neef J, Chepurwar SS, et al. Ca2+ binding to the C2E domain of otoferlin is required for hair cell exocytosis and hearing. Protein Cell. 2024 Apr 1;15(4):305-12.
29. Elliott KL, Pavlínková G, Chizhikov VV, Yamoah EN, Fritzsch B. Development in the Mammalian Auditory System Depends on Transcription Factors. Int J Mol Sci. 2021 Apr 18;22(8):4189.
30. McGovern MM, Groves AK. Specification and plasticity of mammalian cochlear hair cell progenitors. InHair Cell Regeneration. Cham: Springer International Publishing; 2023. p. 105-34.
31. Bouchard M, de Caprona D, Busslinger M, Xu P, Fritzsch B. Pax2 and Pax8 cooperate in mouse inner ear morphogenesis and innervation. BMC Dev Biol. 2010 Aug 20;10:89.
32. Ma Q, Anderson DJ, Fritzsch B. Neurogenin 1 null mutant ears develop fewer, morphologically normal hair cells in smaller sensory epithelia devoid of innervation. J Assoc Res Otolaryngol. 2000 Sep;1(2):129-43.
33. Pauley S, Lai E, Fritzsch B. Foxg1 is required for morphogenesis and histogenesis of the mammalian inner ear. Dev Dyn. 2006 Sep;235(9):2470-82.
34. Fritzsch B, Weng X, Yamoah EN, Qin T, Hui CC, Lebrón-Mora L, Pavlinkova G, et al. Irx3/5 Null Deletion in Mice Blocks Cochlea-Saccule Segregation and Disrupts the Auditory Tonotopic Map. J Comp Neurol. 2024 Dec;532(12):e70008.
35. Filova I, Dvorakova M, Bohuslavova R, Pavlinek A, Elliott KL, Vochyanova S, et al. Combined Atoh1 and Neurod1 Deletion Reveals Autonomous Growth of Auditory Nerve Fibers. Mol Neurobiol. 2020 Dec;57(12):5307-23.
36. Shimojo H, Masaki T, Kageyama R. The Neurog2-Tbr2 axis forms a continuous transition to the neurogenic gene expression state in neural stem cells. Dev Cell. 2024 Aug 5;59(15):1913-23.e6.
37. Tateya T, Imayoshi I, Tateya I, Ito J, Kageyama R. Cooperative functions of Hes/Hey genes in auditory hair cell and supporting cell development. Dev Biol. 2011 Apr 15;352(2):329-40.
38. Li S, He S, Lu Y, Jia S, Liu Z. Epistatic genetic interactions between Insm1 and Ikzf2 during cochlear outer hair cell development. Cell Rep. 2023 May 30;42(5):112504.
39. Bi Z, Ren M, Zhang Y, He S, Song L, Li X, et al. Revisiting the Potency of Tbx2 Expression in Transforming Outer Hair Cells into Inner Hair Cells at Multiple Ages In Vivo. J Neurosci. 2024 Jun 5;44(23):e1751232024.
40. Nakano Y, Wiechert S, Fritzsch B, Bánfi B. Inhibition of a transcriptional repressor rescues hearing in a splicing factor-deficient mouse. Life Sci Alliance. 2020 Oct 21;3(12):e202000841.
41. Koo HY, Oh JH, Durán Alonso MB, Hernández IL, González-Vallinas M, Alonso MT, et al. Analysis of Meis2 knockout mice reveals Sonic hedgehog-mediated patterning of the cochlear duct. Dev Dyn. 2025 Apr;254(4):365-72.
42. McGovern MM, Hosamani IV, Niu Y, Nguyen KY, Zong C, Groves AK. Expression of Atoh1, Gfi1, and Pou4f3 in the mature cochlea reprograms nonsensory cells into hair cells. Proc Natl Acad Sci U S A. 2024 Jan 30;121(5):e2304680121.
43. Oertel D, Cao X-J. The Ventral Cochlear Nucleus. In: Fritzsch B, editor. The Senses: A Comprehensive Reference (Second Edition). Oxford: Elsevier; 2020. p. 517-32.
44. Maricich SM, Xia A, Mathes EL, Wang VY, Oghalai JS, Fritzsch B, et al. Atoh1-lineal neurons are required for hearing and for the survival of neurons in the spiral ganglion and brainstem accessory auditory nuclei. J Neurosci. 2009 Sep 9;29(36):11123-33.
45. Lu H-W, Smith PH, Joris PX. Mammalian octopus cells are direction selective to frequency sweeps by excitatory synaptic sequence detection. Proc Natl Acad Sci U S A. 2022 Nov;119(44):e2203748119.
46. Trussell LO, Oertel D. Microcircuits of the dorsal cochlear nucleus. In: Oliver DL, Cant NB, Fay RR, Popper AN. The Mammalian Auditory Pathways. Cham: Springer International Publishing; 2018. p. 73-99.
47. Mukherjee D, Meng X, Kao JPY, Kanold PO. Impaired Hearing and Altered Subplate Circuits During the First and Second Postnatal Weeks of Otoferlin-Deficient Mice. Cereb Cortex. 2022 Jun 16;32(13):2816-30.
48. Iskusnykh IY, Steshina EY, Chizhikov VV. Loss of Ptf1a leads to a widespread cell-fate misspecification in the brainstem, affecting the development of somatosensory and viscerosensory nuclei. J Neurosci. 2016 Mar 2;36(9):2691-710.
49. Parameshwarappa V, Siponen MI, Watabe I, Karkaba A, Galazyuk A, Noreña AJ. Noise-induced hearing loss alters potassium-chloride cotransporter KCC2 and GABA inhibition in the auditory centers. Sci Rep. 2024 May 9;14(1):10689.
50. Di Bonito M, Studer M. Cellular and molecular underpinnings of neuronal assembly in the central auditory system during mouse development. Front Neural Circuits. 2017 Apr 19;11:18.
51. Milinkeviciute G, Cramer K. The Senses: A Comprehensive Reference. 2020.
52. Eggenschwiler JT, Bulgakov OV, Qin J, Li T, Anderson KV. Mouse Rab23 regulates hedgehog signaling from smoothened to Gli proteins. Dev Biol. 2006 Feb 1;290(1):1-12.
53. Driscoll ME, Tadi P. Neuroanatomy, Inferior Colliculus. 2023 Aug 14. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-.
54. Tran H-N, Nguyen Q-H, Jeong J-e, Loi D-L, Nam YH, Kang TH, et al. The embryonic patterning gene Dbx1 governs the survival of the auditory midbrain via Tcf7l2-Ap2δ transcriptional cascade. Cell Death Differ. 2023 Jun;30(6):1563-74.
55. Jahan I, Kersigo J, Elliott KL, Fritzsch B. Smoothened overexpression causes trochlear motoneurons to reroute and innervate ipsilateral eyes. Cell Tissue Res. 2021 Apr;384(1):59-72.
56. Chumak T, Tothova D, Filova I, Bures Z, Popelar J, Pavlinkova G, et al. Overexpression of Isl1 under the Pax2 Promoter, Leads to Impaired Sound Processing and Increased Inhibition in the Inferior Colliculus. Int J Mol Sci. 2021 Apr 26;22(9):4507.
57. Nakamura H. Midbrain patterning: polarity formation of the tectum, midbrain regionalization, and isthmus organizer. In: Rubenstein J, Chen B, Editors. Patterning and Cell Type Specification in the Developing CNS and PNS. Amsterdam: Academic Press; 2020. p. 87-106.
58. Kim EJ, Hori K, Wyckoff A, Dickel LK, Koundakjian EJ, Goodrich LV, et al. Spatiotemporal fate map of neurogenin1 (Neurog1) lineages in the mouse central nervous system. J Comp Neurol. 2011 May 1;519(7):1355-70.
59. Puelles L, Martínez S, Martínez-De-La-Torre M, Rubenstein JL. Gene maps and related histogenetic domains in the forebrain and midbrain. In: Paxinos G, Editor. The rat nervous system. Amsterdam: Academic Press; 2015. p. 3-24
60. Newman EA, Kim DW, Wan J, Wang J, Qian J, Blackshaw S. Foxd1 is required for terminal differentiation of anterior hypothalamic neuronal subtypes. Dev Biol. 2018 Jul 15;439(2):102-11.
61. Manuel M, Tan KB, Kozic Z, Molinek M, Marcos TS, Razak MFA, et al. Pax6 limits the competence of developing cerebral cortical cells to respond to inductive intercellular signals. PLoS Biol. 2022 Sep 6;20(9):e3001563.
62. Enard W. FOXP2 and the role of cortico-basal ganglia circuits in speech and language evolution. Curr Opin Neurobiol. 2011 Jun;21(3):415-24.
63. Dennis DJ, Han S, Schuurmans C. bHLH transcription factors in neural development, disease, and reprogramming. Brain Res. 2019 Feb 15;1705:48-65.
64. Molnár Z, Luhmann HJ, Kanold PO. Transient cortical circuits match spontaneous and sensory-driven activity during development. Science. 2020 Oct 16;370(6514):eabb2153.
65. Kanold PO, Luhmann HJ. The subplate and early cortical circuits. Annual review of neuroscience. 2010;33:23-48.
66. Goodrich L, Kanold P. Functional circuit development in the auditory system. In: Rubenstein J, Rakic P, Editors. Neural Circuit and Cognitive Development. Amsterdam: Academic Press; 2020. p. 27-55.
67. Yamoah EN, Pavlinkova G, Fritzsch B. The Development of Speaking and Singing in Infants May Play a Role in Genomics and Dementia in Humans. Brain Sci. 2023 Aug 11;13(8):1190.
68. Fritzsch B, Elliott KL, Pavlinkova G. Primary sensory map formations reflect unique needs and molecular cues specific to each sensory system. F1000Research. 2019 Mar 27;8:F1000 Faculty Rev-345.
69. Fritzsch B, Matei VA, Nichols DH, Bermingham N, Jones K, Beisel KW, et al. Atoh1 null mice show directed afferent fiber growth to undifferentiated ear sensory epithelia followed by incomplete fiber retention. Dev Dyn. 2005 Jun;233(2):570-83.