Abstract
Conformational changes of Tau have been described to occur during its fibrillary and non-fibrillary aggregation inside neurons affected in the brain of Alzheimer’s disease (AD) patients. Two consecutive conformations have been described during the progression of the disease: an early conformation detected with the Alz-50 antibody, recognizing Tau molecules folding its amino terminus over its third repeated domain, and a later conformation involving the bending of the proline-rich region over the third repeated domain. α-1-antichymotrypsin (ACT) is an acute phase serum glycoprotein that is overexpressed in the brain of AD cases and associated with extracellular amyloid-ß aggregates. We have recently reported that in a large population of neurons affected in AD brains, Tau protein undergoing the conformational change detected by Tau-66 antibody accumulates as non-fibrillary aggregates and colocalizes with extensive accumulations of granular diffuse intracellular deposits of ACT. In this report, we further analyzed Tau-ACT interactions in the neurons from the hippocampus of AD brains. By using superresolution confocal microscopy and quantitative colocalization analysis, we corroborated the mutual association and mislocalization of conformationally altered Tau protein and ACT to the nuclear compartment. These results suggest that ACT can play an abnormal pathological role in AD by contributing to the abnormal transport of truncated and conformationally altered Tau protein to the nucleus.
Keywords
Alzheimer’s disease, α-1-antichymotrypsin, Confocal microscopy, Nuclear Tau, Tau conformation, Neuropathology
Short Communication
The role of the tau protein under physiological conditions and in association with neurodegenerative processes such as Alzheimer's disease (AD) and other tauopathies differs considerably. Under healthy conditions, Tau participates in the stabilization of microtubules in axonal projections and may also interact with other intracellular components such as the plasma membrane, F-actin, and motor proteins [1]. Moreover, Tau has been detected in the nuclei of neurons and it has been suggested that it may be associated with genetic material and involved in its structural organization [2]. Although in vitro experiments have found that Tau can interact with DNA through its GC-rich or AT-rich DNA regions [3], this information predominantly comes from in vitro experiments using isolated molecules. Extensive data have also been obtained from cell culture systems, both neuronal and non-neuronal, describing a nuclear localization of Tau and alleged involvement in the structural organization of DNA, cell division, and control of gene expression [4]. Tau has been reported from animal models such as transgenic mice as a molecule that protects DNA under physiological and hyperthermia conditions [5]. However, the capability of Tau to locate in the nucleus and participate in additional roles may be a response to the immediate cellular need to ensure its functionality and survival under culture conditions.
Nonetheless, this scenario becomes more complicated when a neuron reaches a structural complexity that allows it to form synaptic connections, as occurs in the CNS, where Tau is predominantly involved in stabilizing the tubulin cytoskeleton [1]. Tau is a molecule susceptible to post-translational modifications, with its level of phosphorylation being the most accepted change controlling its microtubule-binding capacity. Under pathological conditions, such as in AD, Tau turns out to be massively phosphorylated, which reduces its microtubule affinity, leading not only to destabilization of these structures, but also to the gradual accumulation of Tau in the form of abnormal filaments (paired helical filaments) abnormally self-aggregating and the formation of neurofibrillary tangles (NFTs) [1].
Abnormal aggregation of Tau in AD and other tauopathies is the primary cause of neuronal death. Tau also accumulates in dystrophic neurites and in neuropil threads, causing loss of neuronal connectivity in the hippocampus and neocortical areas. However, Tau can also be localized in the nuclear compartment that experiences a high phosphorylation level [6]. This modification reduces its capability to stabilize genetic material and its presumed involvement in the regulation of gene expression. The mechanism by which Tau localizes in the nucleus of AD-affected neurons is unknown. Because the Tau molecule does not contain a canonical nuclear localization sequence (NLS), other mechanisms such as phase separation, binding to nucleoporins, and chaperone transporters may facilitate its abnormal redistribution to the nucleus [7]. We have reported that the Tau molecule also undergoes conformational changes during its abnormal accumulation in the form of paired helical filaments and NFTs, and that these changes seem to follow a continuum depending on the degree of proteolysis of its amino and carboxyl termini [8].
In particular, the Tau-66 antibody recognizes a conformational change that depends on the truncation of its amino and carboxyl termini, and its appearance occurs at intermediate and advanced stages of disease progression. This conformational change is described in Tau-composed NFTs and in granular diffuse aggregates of neurons. However, with the use of Tau-66 antibody, it is also possible to demonstrate conformationally altered Tau in populations of glial cells [9]. These cells are active during AD neurodegeneration in response to many stressful conditions, such as ß-amyloid plaque formation and the resulting induction of oxidative stress. It seems that the expression of Tau protein is also deregulated in glial cells, and although this molecule does not form NFTs similar to those occurring in neurons, Tau can also aggregate in astrocytes and microglial cells in various tauopathies. Various pro-inflammatory cytokines are also overexpressed due to the toxic environment of oxidative stress in the brain parenchyma, leading to abnormal processing of the tau protein and favoring its hyperphosphorylated state [10]. In light of these circumstances, the expression of other factors that modulate inflammation and proteostasis in the brains of AD-affected individuals plays an essential compensatory role. In this regard, acute-phase alpha-1-antichymotrypsin (ACT) is a compensatory factor that reduces the activity of proteases such as chymotrypsin, which is also actively expressed in the brains of AD patients [11]. Astrocytes and microglial cells are one of the primary sources of ACT in the brain and some specific neuronal populations. However, free ACT has undesirable properties that promote in vitro and in situ ß-amyloid aggregation [12]. In cortical neurons cultured in vitro, extracellular exposure to ACT was found to result in increased Tau phosphorylation, which correlated with increased expression of extracellular signal-regulated kinase (ERK) and glycogen synthase kinase-3 (GSK3) [13].
Recently, we found that granular accumulation of ACT in the cytoplasm of hippocampal neurons of AD patients is associated with diffuse Tau protein aggregates that undergo only conformational changes recognized by Tau-66. In an early conformation detected by the Alz-50 antibody, which relies on an intact molecule that preserves several phosphorylated sites at the amino terminus, Tau does not associate with cytoplasmic ACT [14]. This result indicates that ACT may function as a pathological chaperone for Tau, leading to its abnormal aggregation in the cytoplasmic space. We now report that this possible association of conformationally altered Tau and ACT is also found in the nuclear compartment of diverse neuronal populations expressing both proteins (Figure 1). With super-resolution confocal microscopy, we better defined the amorphous and granular nature of the aggregates of both proteins, as well as the evidence for their potential association within the neuronal nucleus (Figure 1). Panoramic images of double-labeling immunofluorescence with Tau-66 and anti-ACT antibodies are shown in panel A in Figure 1, demonstrating the co-expression of both molecules in several neurons in the hippocampus of AD cases. Selected regions of interest in panel A were collected as high-resolution images (Panel B in Figure 1), and better evidence of the presence of Tau and ACT in the nuclear matrix is observed. From the high-resolution of single optical sections (Panel C in Figure 1 and Panel C in Figure 2), we evaluated quantitative colocalization as reported previously [14]. We found that conformationally-altered Tau and ACT colocalize with a significant Person's coefficient of 0.73 (n= 15) (fluorogram in panel D in Figure 2), which may indicate a possible association. Moreover, line scan plots (Panel D in Figure 1) were produced for each fluorescent marker. The scan corresponds to fluorescence intensity along the linear vector drawn in the nuclear space. The plot profiles show that both signals emerge or decrease in synchrony along the distance, which may sum up evidence of possible association. In Figure 2, another neuron showing the same colocalization pattern is projected as 2-D (Panels B and C) and 3-D (Panel A) images to better appreciate the nuclear localization of conformationally altered Tau and ACT.
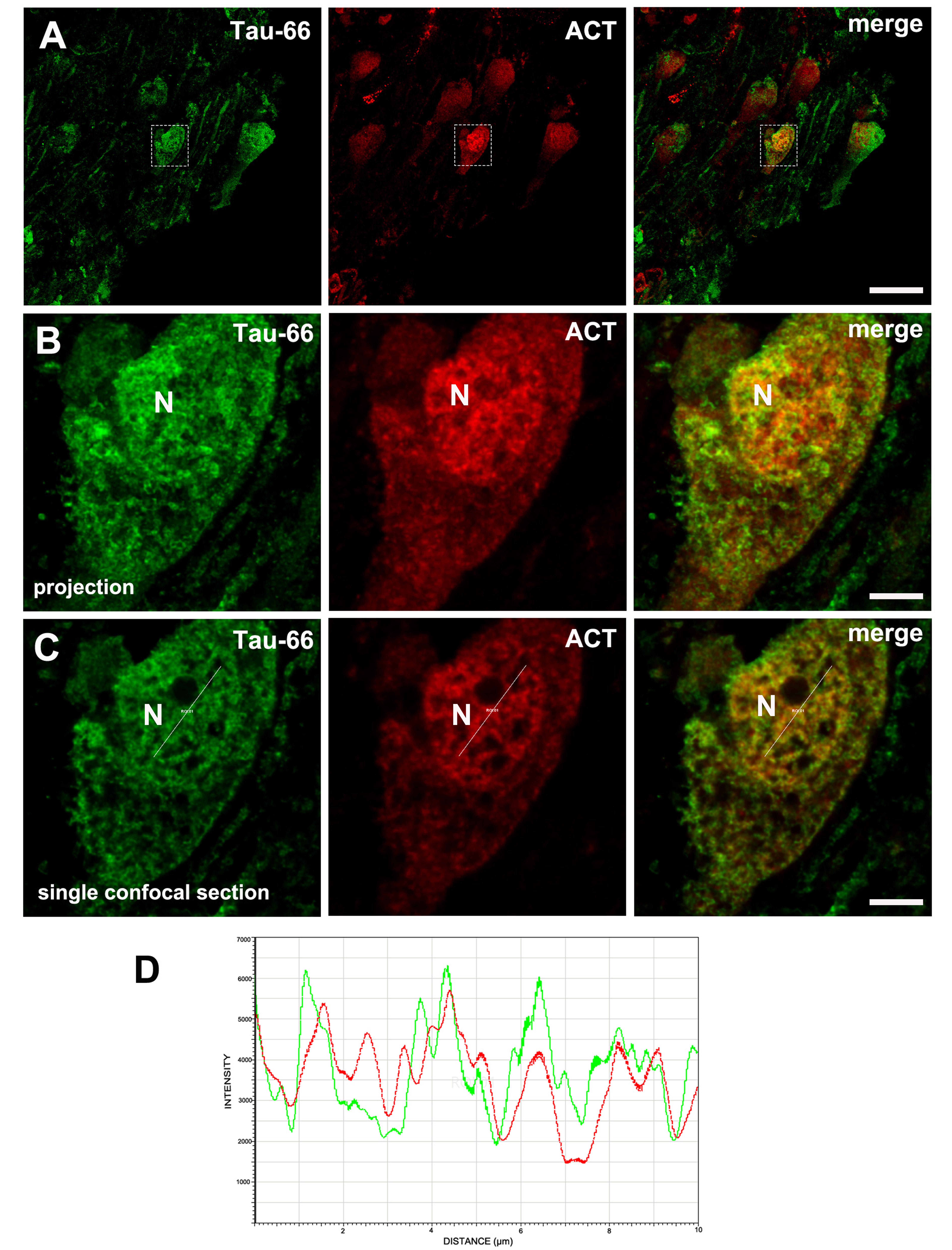
Figure 1. Nuclear distribution and association of Tau with ACT in hippocampal neurons in AD brains. Double labeling immunofluorescence with Tau-66 (to recognize conformation change in Tau molecule) and polyclonal anti-ACT antibodies. (A) Panoramic view of the CA1 sector. High-resolution images from the region of interest in (A) are depicted in panel (B). (C) A single optical section was selected to analyze quantitative colocalization under super-resolution confocal microscopy. (D) The line scan plot shows the fluorescence intensity of each fluorochrome along the lines drawn in the nuclear area of the cell in panel (C). N: nucleus. Scale bars: 23 µm in (A), 5 µm in (B) and (C).
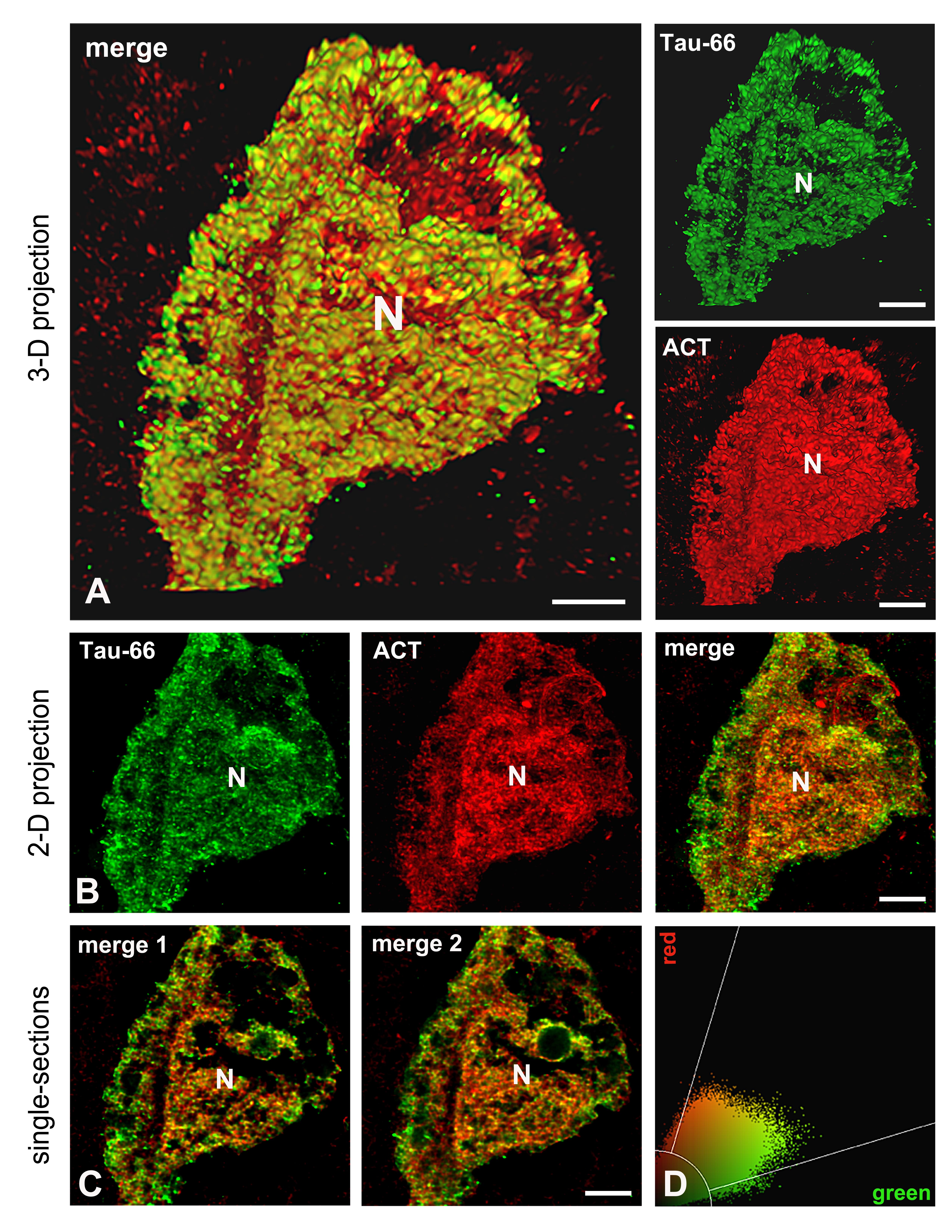
Figure 2. 2-D and 3-D reconstruction of neuronal aggregates of conformationally altered Tau protein and ACT. In (A), the 3-D reconstruction of images displayed in B shows the close association between Tau and ACT in the cytoplasm and nucleus of the cell. From 2-D projections (B), single optical sections were obtained (C), and their fluorograms (D) were used to analyze the significance of colocalization through Pearson´s analysis. N: nucleus. Scale bars: 3 µm in merge, and 4 µm in the green and red channels of (A). 4 µm in (B) and (C).
Most of these neuronal populations did not seem to present severe morphological changes and no well-developed NFTs were seen in the cytoplasm, possibly indicating that this phenomenon occurs in neurons that have not yet entered an advanced neurodegenerative process. Nevertheless, the Tau molecule becomes truncated and conformationally altered, making it more susceptible to interaction with other pathological chaperones that transport Tau to the nucleus. On the other hand, deregulation of ACT expression also occurs in other pathological processes such as cancer. Although the mechanism is still unknown, ACT is thought to contribute to the deregulation of factors associated with the control of genes that promote cell malignancy [15].
We still cannot say that Tau and ACT contribute to altering the structure and expression of genetic material in AD-affected neurons. However, their conformational and post-translational modifications may favor their abnormal permanence in this compartment with pathological consequences. These two molecules may be part of a scaffold of factors that accumulate in the nucleus, favoring or altering the action of various transcription factors, leading to subsequent deterioration of the neuron functioning. From these results, it is worthwhile to use cell and animal models to characterize the consequences of the synergistic accumulation of Tau and ACT in the nuclear compartment as an alternative mechanism to Tau toxicity that may occur in AD-affected neurons.
Conclusion
In summary, our observation supports the evidence that Tau, under pathological conditions, undergoes conformational changes that favor its abnormal localization to the nuclear component, perhaps promoted or stabilized by other chaperone molecules such as ACT. This action could be another pathologic feature of Tau undergoing abnormal processing and intracellular mislocalization in AD-affected neurons long before the occurrence of fibrillary aggregates.
Acknowledgments
The authors thank Dr. Lester I. Binder for sharing Tau-66 antibody. Confocal microscopy facilities were provided by the Confocal Microscopy Unit at the Cell Biology Department of CINVESTAV-IPN (CONAHCyT-Mexico grant: 300062). We thank Daniela Ripova and Zdena Kristofikova for their help to have access to human brain tissue at NUDZ. Proofread by Modern Manuscript Editing Services.
Author Contributions
Study conception: Perla H. Horta-López, Jan Rícny, Benjamín Florán Garduño and Francisco Garcia-Sierra. Acquisition of data: Perla H. Horta-López and Francisco Garcia-Sierra. Analysis and interpretation of data: Perla H. Horta-López, Jan Rícny, Benjamín Florán Garduño and Francisco Garcia-Sierra. Drafting of the manuscript: Jan Rícny, Benjamín Florán Garduño and Francisco Garcia-Sierra. All authors approved the final version of the manuscript to be published.
Conflict of Interest Statement
The authors declare no conflict of interest for this article.
Ethics Statement
All experiments were performed in accordance with the Declaration of Helsinki, and the study was formally approved by the local ethics committee of the Prague Psychiatric Center and in compliance with Laws No. 129–2003 and 130–2003.
Funding Information
Supported by Consejo Nacional de Humanidades, Ciencias y Tecnologías de Mexico (CONAHCYT-Mexico) (grant 255224 to Francisco Garcia-Sierra, and scholarship 778468 to Perla H. Horta-López).
References
2. Sultan A, Nesslany F, Violet M, Bégard S, Loyens A, Talahari S, et al. Nuclear tau, a key player in neuronal DNA protection. J Biol Chem. 2011;286:4566-75.
3. Sjoberg MK, Shestakova E, Mansuroglu Z, Maccioni RB, Bonnefoy E. Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci. 2006;119:2025–2034.
4. Galas MC, Bonnefoy E, Buee L, Lefebvre B. Emerging Connections Between Tau and Nucleic Acids. In: Takashima A, Wolozin B, Buee L, eds. Advances in Experimental Medicine and Biology. Tau Biology. Singapore: Springer, 2019:135-143.
5. Violet M, Delattre L, Tardivel M, Sultan A, Chauderlier A, Caillierez R, et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Frontiers in Cellular Neuroscience. 2014;8:84e.
6. Maina MB, Al-Hilaly YK, Serpell LC. Nuclear Tau and Its Potential Role in Alzheimer's Disease. Biomolecules. 2016;6:9.
7. Diez Lisa, Susanne Wegmann S. Nuclear Transport Deficits in Tau-Related Neurodegenerative Diseases. Frontiers in Neurology 2020;11:1056.
8. Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW, Binder LI. Conformational changes and truncation of tau protein during tangle evolution in Alzheimer's disease. J Alzheimers Dis. 2003;5(2):65-77.
9. Ghoshal N, Garcia-Sierra F, Fu Y, Beckett LA, Mufson EL, Kuret J, et al. Tau-66: evidence for a novel tau conformation in Alzheimer's disease. J Neurochem. 2001;77:1372-85.
10. Bai R. Oxidative stress: The core pathogenesis and mechanism of Alzheimer's disease. Ageing Research Reviews. 2022;77:101619.
11. Mezer M, Rogaliński J, Przewoźny S, Chojnicki M, Niepolski L, Sobieska M, et al. SERPINA3: Stimulator or Inhibitor of Pathological Changes. Biomedicines 2023;11:156.
12. Abraham CR, Shirahama T, Potter H. Alpha 1-antichymotrypsin is associated solely with amyloid deposits containing the beta-protein. Amyloid and cell localization of alpha 1-antichymotrypsin. Neurobiol Aging. 1990;11:123-9.
13. Padmanabhan J, Levy M, Dickson DW, Potter H. Alpha1- antichymotrypsin, an inflammatory protein overexpressed in Alzheimer's disease brain, induces tau phosphorylation in neurons. Brain. 2006;129(11):3020-34.
14. Horta-Lopez P, Mendoza-Franco G, Rodriguez-Cruz F, Torres-Cruz FM, Hernámdez-Echeagaray E, Jarero-Basulto JJ, et al. Association of a-1-Antichymotrypsin Expression with the Development of Conformational Changes of Tau Protein in Alzheimer’s Disease Brain. Neuroscience. 2023;518:83-100.
15. Soman A, Nair A. Unfolding the cascade of SERPINA3: Inflammation to cancer. BBA - Reviews on Cancer. 2022;1877:188760.