Abstract
Pediatric nonalcoholic fatty liver disease (NAFLD) affects 1 in 10 children in the US, increases risk of cirrhosis and transplantation in early adulthood, and shortens lifespan, even after transplantation. Exposure to maternal obesity and/or a diet high in fat, sugar and cholesterol is strongly associated with development of NAFLD in offspring. However, mechanisms by which “priming” of the immune system in early life increases susceptibility to NAFLD are poorly understood. Recent studies have focused on the role “non-reparative” macrophages play in accelerating inflammatory signals promoting fibrogenesis. In this Commentary, we review evidence that the pioneering gut bacteria colonizing the infant intestinal tract remodel the naïve immune system in the offspring. Epigenetic changes in hematopoietic stem and progenitor cells, induced by exposure to an obesogenic diet in utero, may skew lineage commitment of myeloid cells during gestation. Further, microbial dysbiosis in neonatal life contributes to training innate immune cell responsiveness in the gut, bone marrow, and liver, leading to developmental programming of pediatric NAFLD. Comprehensive understanding of how different gut bacteria and their byproducts shape development of the early innate immune system and microbiome will uncover early interventions to prevent NAFLD pathophysiology.
Keywords
Pediatric NAFLD, Trained immunity, Pioneering bacteria, Microbiome, Epigenetic reprogramming, Hematopoietic stem cells
Pediatric NAFLD and Why It Is a Pressing Issue
Nonalcoholic fatty liver disease (NAFLD), a spectrum of pathologies ranging from simple steatosis to fibrosis and cirrhosis, is the most common cause of chronic liver disease, affecting over 80% of adults with obesity [1], one third of obese children ages 3-18 in North America [2] and ~10% of the general pediatric population [2]. NAFLD in children progresses more rapidly than in adults [3-7], often leading to cirrhosis and transplantation in early adulthood [6]. Half of children presenting with NAFLD have already progressed to the more serious form of nonalcoholic steatohepatitis (NASH) at time of diagnosis [2,8,9] and their survival, even after transplantation, is shortened when compared with the general population [5]. Maternal obesity is a significant risk factor for pediatric NAFLD [10,11]. However, a major limitation in this field is the lack of fundamental understanding as to how maternal diet and/or obesity sets liver physiology and development of the immune system, beginning early in life, on a course toward NAFLD.
NASH is characterized by inflammation, oxidative stress, mitochondrial dysfunction, elevated levels of pro-inflammatory cytokines and fibrosis. Data from our studies in a nonhuman primate model of maternal obesity [10,12-18], combined with findings from other studies in mice [19] and humans [20], indicate that risk factors for NAFLD begin in utero, altering tissue function at the cellular and molecular levels. Persistence of liver steatosis and inflammation in juvenile animals switched to a healthy diet at weaning suggests that developmental changes have permanent epigenetic effects which alter metabolic outcomes and increase vulnerability to accelerated fibrosis in offspring [10,11,21]. DNA methylation, covalent modification of histones, and the expression of non-coding RNA are epigenetic phenomena found in livers from children [22-25], adults [26-33], and rodents [34-39] with NAFLD (reviewed recently by Campisano, et al. [40]). Still, insights into the mechanisms by which maternal diet and obesity prime the immune system toward inflammation and liver damage are lacking.
In NAFLD, portal infiltration of macrophages is an early event predicting disease progression, and occurs in the steatotic liver before inflammation or fibrosis develops [41]. Hepatic inflammation is driven, in part, by activated endogenous liver macrophages (Kupffer cells), innate immune cells arising from fetal liver, and from infiltrating monocyte/macrophages arising from bone marrow precursors [42,43]. Kupffer cells and monocyte/ macrophages can either promote hepatic inflammation and fibrosis (M1-like, pro-inflammatory) [10,44-54], or resolve inflammation and prevent progression to fibrosis (M2-like, pro-restorative/reparative) [55]. In children with NASH, numerous activated macrophages are found in the spaces between damaged hepatocytes [56]. We (and others) have shown that mice exposed to a maternal “Western-style” diet (WD) have increased pro-inflammatory macrophage activation in the liver and accelerated hepatic fibrosis as adults [57]. Further, our published data in a nonhuman primate model demonstrated increased expression of pro-inflammatory cytokines (IL1B and TNFA) in hepatic macrophages isolated from 1-year-old offspring born to WD-fed mothers and weaned to a chow diet [14]. Whether maternal diets during gestation or lactation alter the fate of developing macrophage precursors remains an important unanswered question.
Gut Microbial Dysbiosis in Early Life Influences the Developing Immune System
Evidence suggests maternal and postnatal factors that impact the developing infant gut microbiome include maternal diet [58], infant diet in early life [59], antibiotic use, and delivery by Caesarean section [60,61]. Studies in animals [62-65] and neonates [66,67] demonstrate that “pioneering” gut bacteria in early life profoundly shape development of the innate and adaptive branches of the immune system, with persistent effects on immune function later in life [68,69]. Because both the gut microbiome and gut immune cells develop and mature during the neonatal period [70,71], even a brief disruption to the microbial community structure during this window can induce immunological changes that persist into adulthood [69]. For example, Olszak, et al. showed colonizing neonatal - but not adult - germ-free mice with pioneering microbes protects from immune cell-mediated pathology in adulthood [69]. Together, these studies demonstrate the critical importance of initial colonizers to the gut microbiome – immunity axis [72].
Most microbiota-driven immune alterations are assumed to be postnatal effects induced by the neonate’s own microbiota [73-75]. However, detection of bacteria in the placenta, amniotic fluid, and meconium suggests the possibility of fetal colonization during gestation, which alters development of the naïve fetal innate immune system [76] and impacts maturation of the hematopoietic system [77]. Gomez de Agüero, et al. showed this by transiently colonizing germ-free pregnant dams with Escherischia coli, which led to enhanced ILC3 and F4/80+ CD11c+ mononuclear cell populations in the gut of neonates, reprogrammed intestinal transcriptional profiles, and increased expression of genes involved in metabolism, oxidative stress, and innate immunity [78]. Moreover, bacterial colonization in early life influences immune development in primary lymphoid organs beyond the gut, including in the bone marrow [77]. Gut microbial and nonmicrobial ligands, including short-chain fatty acids (SCFA) and bile acids, induce a memory response in innate cells mediated by pattern recognition receptors (PRRs), which recognize microbe- or pathogen-associated molecular patterns (MAMPs or PAMPs) [79]. Pattern recognition receptors include families of Toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)– like receptors (NLRs). Microbe- and pathogen-associated molecular pattern recognition via these PRRs affects differentiation and function of myeloid and lymphoid lineage immune cells [80-82], inducing both myeloid bias in long-term hematopoietic stem cells within the bone marrow [83] and immune memory in differentiated descendents [84]. Lipopolysaccharide (LPS), a canonical ligand for TLR, dose- and duration-dependently induces tolerance or potentiates innate immune memory [85]. LPS isolated from E. coli has immunostimulatory activity leading to endotoxin tolerance, which is inhibited by LPS from Bacteroides [86], supporting the hypothesis that in early life, the abundance ratio of Enterobacteriaceae to other commensals critically regulates immune development and health in later life.
In addition to bacteria, bacteria-derived dietary metabolites transfer from mother to fetus, affecting immune development. In mice, feeding pregnant dams dietary fiber or acetate alone protected offspring from allergic airway inflammation [87] and maternally-derived SCFAs played a role in Foxp3+ regulatory T cell generation in the neonatal thymus [88]. Maternal gut bacterial products other than SCFAs may have effects on programming immunity in the offspring. Metabolites including taurine, polyamines, retinoic acid, and indoles (byproducts of tryptophan catabolism) have roles in maintaining immune homeostasis, gut barrier integrity, and arginine levels, as well as regulating the inflammasome [89].
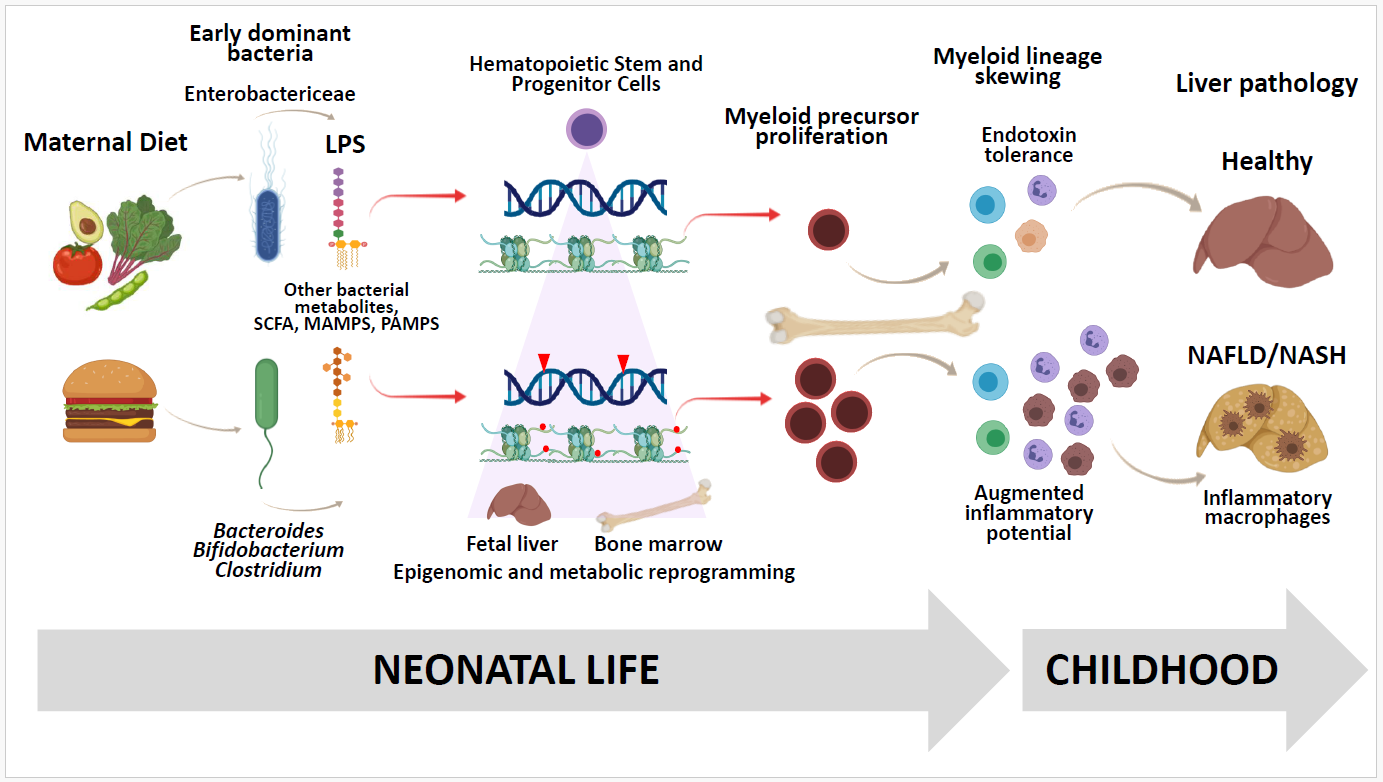
Enterobacteriaceae are an important family of Gammaproteobacteria, a class of aerobic, LPS-producing pioneering microbes that are abundant in the stool from full-term, vaginally-delivered newborns [90]. E. coli and Enterobacter, facultative anaerobic pioneering bacteria, rapidly grow and metabolize lactose from breast milk to acetate and other SCFAs and create a reduced acidic environment. This environment is favorable for later colonization by slower-growing, anaerobic acidophiles such as Bacteroides, Bifidobacterium, and Clostridium [91,92], which consume most of the available sugar and excrete large amounts of acetate and other SCFAs [93]. Unlike in adults, early exposure to Enterobacteriaceae in newborn rodents and humans provides LPS-driven inflammatory challenges important for training or “priming” the early immune system, and protecting against excessive inflammatory, autoimmune, and metabolic disorders later in life. LPS/endotoxin binds to receptors on innate immune cells, including TLR4, and modulates the host innate immune response though mechanisms such as endotoxin tolerance or trained immunity [94-96]. Subtypes derived from specific Bacteroides species exhibit lower endotoxicity than LPS isolated from other enteric bacteria [97]. Immune sequelae linked to early life dysregulation of the Enterobacteriaceae to Bacteroidetes balance were recently described in human infants. Vatanen, et al. elegantly showed Bacteroides species, in the microbiota of infants from countries with high susceptibility to allergies and type 1 diabetes (T1D), produced an LPS subtype that inhibited immunostimulatory activity of E. coli LPS in vitro, compared with those infants colonized predominantly by Enterobacteriaceae. In vivo, intraperitoneal injection of E. coli-derived LPS led to endotoxin tolerance in immune cells and delayed onset of T1D in a mouse model, whereas LPS from Bacteroides was not protective [86].
The impact of maternal obesity on pioneering bacteria in infants was studied by Lemas, et al. [98] who found reduced abundance of Gammaproteobacteria in stool from 2-week-old infants born to obese mothers, when compared with microbiota from infants born to normal weight mothers [99]. Soderborg, et al. colonized germfree mice with microbes from infants born to obese mothers, and these mice, when challenged with a shortterm postnatal WD, exhibited elevated markers of inflammation and endoplasmic reticulum stress in liver, as well as accelerated obesity and NAFLD. Moreover, these animals exhibited dampened LPS-induced inflammation in bone marrow-derived macrophages (BMDMs) and impaired phagocytosis [99]. A unique feature of pediatric NAFLD is the predilection for children to deposit fat and develop inflammation in the periportal region vs. the more classic perivenular distribution seen in adults [4,100]. This difference is poorly understood, but clinically relevant because periportal inflammation is associated with advanced liver disease [101]. Germ-free mice colonized with microbiota from infants exposed to maternal obesity showed histological evidence for increased periportal inflammation, even while consuming a control chow diet [99]. These provocative findings suggest a mechanistic role for the early life gut microbiota in priming innate immune dysfunction prior to the development of childhood obesity.
Dietary Exposures and Epigenetic Rewiring Skew Hematopoiesis to Promote Chronic Inflammation
The fetal liver is the major hematopoietic organ of the developing immune system. During gestation, the fetal liver is seeded with monocytes, which are progenitors of liver resident Kupffer cells [102,103], and hematopoietic stem and progenitor cells (HSPCs) which migrate to the bone marrow, where most stay for the remainder of life [104]. HSPCs in the bone are capable of producing all blood cells of the lymphoid (adaptive immune) and myeloid (innate immune, erythroid, platelets) lineages, including monocytes and their macrophage descendants. HSPC-derived monocytes in the fetal liver give rise to Kupffer cells, which maintain self-renewing capabilities throughout life [102,105]. By contrast, infiltrating tissue macrophages differentiate from monocytes that are continuously generated from HSPCs in the marrow and recruited to the liver by damage signals (monocyte/ macrophages) [106]. Diet-induced gut microbial dysbiosis shapes development and function of the immune system, in part by regulating the differentiation of HSPCs [107]. Intriguingly, maternally-derived HSPCs have been detected in cord blood [108]. Whether these maternal cells, programmed by a poor diet, seed the fetal bone marrow and educate the developing immune system, or whether an obesogenic maternal diet and resultant gut microbial dysbiosis directly program neonatal HSPCs to promote NAFLD are questions warranting further research.
Myeloid cells (monocytes/macrophages), innate lymphoid cells (including NK cells), and bone marrow progenitors [109] exhibit innate immune memory. This memory involves epigenetic rewiring after an initial inflammatory insult and a rapid, non-specific enhanced response to subsequent exposures [109]. Microbial signals, including peptidoglycans [110], Bacillus Calmette-Guérin [111], and β-glucan [83], alter epigenetic modifications in HSPCs, induce long-lasting changes in cellular lineages, and stimulate inflammatory priming of differentiated myeloid cells [77]. The induced memory may persist from weeks to months [112,113]. In fetal mice, exposure to maternal WD remodels fetal liver HSPCs to exacerbate the inflammatory immune response, skew commitment to the myeloid lineage, and favor differentiation at the expense of self-renewal [114]. Later-life consequences of this early adaptation can be observed in adult WDfed mice, where HSPCs are biased toward the myeloid lineage and generate a large pool of pro-inflammatory cells [115]. Moreover, in response to short-term post-natal WD exposure, Christ, et al. found upregulation of genes involved in cellular proliferation, skewing of granulocytemonocyte progenitors toward the monocytic cell lineage, and increased availability of enhancer regions (including TLR4) [116].
In our mouse model of maternal WD, we showed elevated fumarate in liver macrophages and BMDMs from adult offspring of obese pregnancy [117]. This metabolic change triggers epigenetic remodeling toward a pro-inflammatory phenotype. Innate immune memory is characterized by a potentiated response to inflammatory stimuli, accompanied by a metabolic shift to aerobic glycolysis and a dysfunctional TCA cycle, termed “metabolic reprogramming” [118]. Metabolic and epigenetic reprogramming occurs in myeloid cells [119,120], including HSPCs [83] and their descendants, and alters transcription of genes in inflammatory, immune, and metabolic pathways [116]. TCA cycle intermediates, such as succinate and fumarate, promote expression of genes supporting the pro-inflammatory (M1-like) macrophage phenotype through stabilization of hypoxia-inducible factor-1α [121-123] and histone acetylation of glycolytic enzyme genes, including hexokinase 2 and lactate dehydrogenase [124]. Moreover, α-ketoglutarate increases expression of genes promoting a reparative (M2-like) phenotype through epigenetic regulation [125,126].
Finally, Wang, et al. showed that DNA hypermethylation at the peroxisome proliferator-activated receptor γ1 promoter in adipose tissue macrophages suppressed the ability of these cells to adopt an alternatively activated, reparative phenotype (characterized by elevated “M2” markers such as ARG1, MRC1, and CLEC10A). Moreover, failure to adopt an M2-like phenotype was associated with weight gain and insulin resistance in mice fed a chronic high-fat diet [127]. These studies suggest that HSPC immunometabolism, even in early life, contributes to programming adult metabolic disease when dysregulated by exposure to an obesogenic maternal diet. It will be important for future work to determine whether similar mechanisms act in bone marrow and the liver.
In conclusion, pediatric NAFLD is a growing problem worldwide with a complex pathophysiology. Therefore, it is critical to elucidate factors driving development of the neonatal immune system, particularly in bone marrow and liver, to determine how maternal obesity alters infant immunity and drives development of pediatric NAFLD. Mechanistic studies, including perinatal maternal or infant supplementation with specific bacteria, could identify bacterial strains or metabolic functions responsible for pro-inflammatory priming of the early innate immune system. Going forward, it will be important to advance our knowledge on how the immune system senses changes in early dietary metabolite composition to either initiate an appropriate inflammatory response or promote inflammatory pathophysiology, such as NAFLD. We must 1) determine whether dietary metabolites other than maternal SCFAs are transferred to the fetus in utero, or to offspring in early life, and characterize metabolite effects on development of the immune system in the fetal liver and/or offspring bone marrow; and 2) evaluate the impact of maternal metabolites on colonization and development of the infant intestinal microbiota and test if these early bacteria exert life-long epigenetic changes in HSPCs and their macrophage descendants, accelerating disease pathophysiology. Comprehensive understanding of how maternal diet and obesity influence development of the innate immune system continues to be a major challenge. But, hopefully, this work will lead to early interventions to prevent numerous metabolic diseases associated with inflammation, including obesity, cardiovascular disease, and NAFLD pathophysiology in children.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Sources of Funding
This study was supported by the National Institutes of Health grant NIH/NIDDK 1R01DK121951-01A1 (KJ, JF) and Presbyterian Health Foundation/Harold Hamm Diabetes Center Bridge Grant (KJ).
Authors Contributions
All authors contributed to writing and editing the manuscript. All authors approved the final version.
Acknowledgements
Not applicable. Only the authors listed on the manuscript contributed to the article.
References
2. Anderson EL, Howe LD, Jones HE, Higgins JP, Lawlor DA, Fraser A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: a systematic review and meta-analysis. PloS One. 2015 Oct 29;10(10):e0140908.
3. Vos MB, Abrams SH, Barlow SE, Caprio S, Daniels SR, Kohli R, et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). Journal of Pediatric Gastroenterology and Nutrition. 2017 Feb;64(2):319.
4. Crespo M, Lappe S, Feldstein AE, Alkhouri N. Similarities and differences between pediatric and adult nonalcoholic fatty liver disease. Metabolism. 2016 Aug 1;65(8):1161-71.
5. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non-alcoholic fatty liver disease in children: a follow-up study for up to 20 years. Gut. 2009 Nov 1;58(11):1538-44.
6. Doycheva I, Issa D, Watt KD, Lopez R, Rifai G, Alkhouri N. Nonalcoholic steatohepatitis is the most rapidly increasing indication for liver transplantation in young adults in the United States. Journal of Clinical Gastroenterology. 2018 Apr 1;52(4):339-46.
7. Carter-Kent C, Brunt EM, Yerian LM, Alkhouri N, Angulo P, Kohli R, et al. Relations of steatosis type, grade, and zonality to histological features in pediatric nonalcoholic fatty liver disease. Journal of Pediatric Gastroenterology and Nutrition. 2011 Feb 1;52(2):190-7.
8. Goyal NP, Schwimmer JB. The progression and natural history of pediatric nonalcoholic fatty liver disease. Clinics in Liver Disease. 2016 May 1;20(2):325-38.
9. Brumbaugh DE, Friedman JE. Developmental origins of nonalcoholic fatty liver disease. Pediatric Research. 2014 Jan;75(1):140-7.
10. Wesolowski SR, El Kasmi KC, Jonscher KR, Friedman JE. Developmental origins of NAFLD: a womb with a clue. Nature Reviews Gastroenterology & Hepatology. 2017 Feb;14(2):81.
11. Ayonrinde OT, Oddy WH, Adams LA, Mori TA, Beilin LJ, de Klerk N, et al. Infant nutrition and maternal obesity influence the risk of non-alcoholic fatty liver disease in adolescents. Journal of Hepatology. 2017 Sep 1;67(3):568- 76.
12. McCurdy CE, Bishop JM, Williams SM, Grayson BE, Smith MS, Friedman JE, et al. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. The Journal of Clinical Investigation. 2009 Feb 2;119(2):323-35.
13. Wesolowski SR, Mulligan CM, Janssen RC, Baker II PR, Bergman BC, D’Alessandro A, et al. Switching obese mothers to a healthy diet improves fetal hypoxemia, hepatic metabolites, and lipotoxicity in non-human primates. Molecular Metabolism. 2018 Dec 1;18:25-41.
14. Thorn SR, Baquero KC, Newsom SA, El Kasmi KC, Bergman BC, Shulman GI, et al. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes. 2014 Aug 1;63(8):2702-13.
15. McCurdy CE, Schenk S, Hetrick B, Houck J, Drew BG, Kaye S, et al. Maternal obesity reduces oxidative capacity in fetal skeletal muscle of Japanese macaques. JCI Insight. 2016 Oct 6;1(16).
16. Suter MA, Chen A, Burdine MS, Choudhury M, Harris RA, Lane RH, et al. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. The FASEB Journal. 2012 Dec;26(12):5106-14.
17. Campodonico-Burnett W, Hetrick B, Wesolowski SR, Schenk S, Takahashi DL, Dean TA, et al. Maternal Obesity and Western-style Diet Impair Fetal and Juvenile Offspring Skeletal Muscle Insulin-Stimulated Glucose Transport in Nonhuman Primates. Diabetes. 2020 Apr 30;69(7):1389-1400.
18. Grant WF, Nicol LE, Thorn SR, Grove KL, Friedman JE, Marks DL. Perinatal exposure to a high-fat diet is associated with reduced hepatic sympathetic innervation in one-year old male Japanese macaques. PloS One. 2012 Oct 30;7(10):e48119.
19. Thompson MD, Cismowski MJ, Trask AJ, Lallier SW, Graf AE, Rogers LK, et al. Enhanced steatosis and fibrosis in liver of adult offspring exposed to maternal high-fat diet. Gene Expression The Journal of Liver Research. 2016 Aug 31;17(1):47-59.
20. Newton KP, Feldman HS, Chambers CD, Wilson L, Behling C, Clark JM, et al. Low and high birth weights are risk factors for nonalcoholic fatty liver disease in children. The Journal of Pediatrics. 2017 Aug 1;187:141-6.
21. Nash MJ, Frank DN, Friedman JE. Early microbes modify immune system development and metabolic homeostasis—the “restaurant” hypothesis revisited. Frontiers in Endocrinology. 2017 Dec 13;8:349.
22. Geurtsen ML, Jaddoe VW, Salas LA, Santos S, Felix JF. Newborn and childhood differential DNA methylation and liver fat in school-age children. Clinical Epigenetics. 2020 Dec;12(1):1-0.
23. Oses M, Margareto Sanchez J, Portillo MP, Aguilera CM, Labayen I. Circulating miRNAs as biomarkers of obesity and obesity-associated comorbidities in children and adolescents: a systematic review. Nutrients. 2019 Dec;11(12):2890.
24. Brandt S, Roos J, Inzaghi E, Kotnik P, Kovac J, Battelino T, et al. Circulating levels of miR-122 and nonalcoholic fatty liver disease in pre-pubertal obese children. Pediatric Obesity. 2018 Mar;13(3):175-82.
25. Wang S, Song J, Yang Y, Zhang Y, Wang H, Ma J. HIF3A DNA methylation is associated with childhood obesity and ALT. PloS One. 2015 Dec 30;10(12):e0145944.
26. Lai Z, Chen J, Ding C, Wong K, Chen X, Pu L, et al. Association of Hepatic Global DNA Methylation and Serum One-Carbon Metabolites with Histological Severity in Patients with NAFLD. Obesity. 2020 Jan;28(1):197-205.
27. Fernández-Tussy P, Fernández-Ramos D, Lopitz- Otsoa F, Simón J, Barbier-Torres L, Gomez-Santos B, et al. miR-873-5p targets mitochondrial GNMT-Complex II interface contributing to non-alcoholic fatty liver disease. Molecular Metabolism. 2019 Nov 1;29:40-54.
28. Yu F, Wang X, Zhao H, Hao Y, Wang W. Decreased Serum miR-1296 may Serve as an Early Biomarker for the Diagnosis of Non-Alcoholic Fatty Liver Disease. Clinical Laboratory. 2019 Oct 1;65(10).
29. Wu J, Zhang R, Shen F, Yang R, Zhou D, Cao H, Chen G, Pan Q, Fan J. Altered DNA methylation sites in peripheral blood leukocytes from patients with simple steatosis and nonalcoholic steatohepatitis (NASH). Medical Science Monitor: International Medical Journal of Experimental and Clinical Research. 2018;24:6946.
30. Loomba R, Gindin Y, Jiang Z, Lawitz E, Caldwell S, Djedjos CS, et al. DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight. 2018 Jan 25;3(2):e96685.
31. Pirola CJ, Gianotti TF, Castaño GO, Mallardi P, San Martino J, Ledesma MM, et al. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 2015 May 1;64(5):800-12.
32. Ahrens M, Ammerpohl O, von Schönfels W, Kolarova J, Bens S, Itzel T, et al. DNA methylation analysis in nonalcoholic fatty liver disease suggests distinct diseasespecific and remodeling signatures after bariatric surgery. Cell Metabolism. 2013 Aug 6;18(2):296-302.
33. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, et al. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013 Nov 1;145(5):1076-87.
34. Liang Q, Chen H, Xu X, Jiang W. miR-182- 5p attenuates high-fat-diet-induced nonalcoholic steatohepatitis in mice. Annals of Hepatology. 2019 Jan 23;18(1):116-25.
35. Yang YL, Kuo HC, Wang FS, Huang YH. MicroRNA- 29a Disrupts DNMT3b to ameliorate diet-induced nonalcoholic steatohepatitis in mice. International Journal of Molecular Sciences. 2019 Jan;20(6):1499.
36. Gutierrez Sanchez LH, Tomita K, Guo Q, Furuta K, Alhuwaish H, Hirsova P, et al. Perinatal nutritional reprogramming of the epigenome promotes subsequent development of nonalcoholic steatohepatitis. Hepatology Communications. 2018 Dec;2(12):1493-512.
37. Li YY, Tang D, Du YL, Cao CY, Nie YQ, Cao J, et al. Fatty liver mediated by peroxisome proliferator-activated receptor-a DNA methylation can be reversed by a methylation inhibitor and curcumin. Journal of Digestive Diseases. 2018 Jul;19(7):421-30.
38. Wankhade UD, Zhong Y, Kang P, Alfaro M, Chintapalli SV, Thakali KM, et al. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS One. 2017 Apr 17;12(4):e0175675.
39. Pruis MG, Lendvai A, Bloks VW, Zwier MV, Baller JF, De Bruin A, et al. Maternal western diet primes nonalcoholic fatty liver disease in adult mouse offspring. Acta Physiologica. 2014 Jan;210(1):215-27.
40. Campisano S, La Colla A, Echarte SM, Chisari AN. Interplay between early-life malnutrition, epigenetic modulation of the immune function and liver diseases. Nutrition Research Reviews. 2019 Jun 1;32(1):128-45.
41. Gadd VL, Skoien R, Powell EE, Fagan KJ, Winterford C, Horsfall L, et al. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology. 2014 Apr;59(4):1393-405.
42. Baffy G. Kupffer cells in non-alcoholic fatty liver disease: the emerging view. Journal of Hepatology. 2009 Jul 1;51(1):212-23.
43. Carpino G, Nobili V, Renzi A, De Stefanis C, Stronati L, Franchitto A, et al. Macrophage activation in pediatric nonalcoholic fatty liver disease (NAFLD) correlates with hepatic progenitor cell response via Wnt3a pathway. PLoS One. 2016 Jun 16;11(6):e0157246. 321
44. Kasmi KC, Stenmark KR. Contribution of metabolic reprogramming to macrophage plasticity and function. In Seminars in Immunology. 2015;27(4):267-275.
45. Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nature Reviews Immunology. 2014 Mar;14(3):181-94.
46. Lade A, Noon LA, Friedman SL. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Current Opinion in Oncology. 2014 Jan;26(1):100.
47. Ramachandran P, Iredale JP. Macrophages: central regulators of hepatic fibrogenesis and fibrosis resolution. Journal of Hepatology. 2012 Jun 1;56(6):1417-9.
48. Tilg H, Moschen AR, Szabo G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology. 2016 Sep;64(3):955-65.
49. Kisseleva T, Brenner DA. The phenotypic fate and functional role for bone marrow-derived stem cells in liver fibrosis. Journal of Hepatology. 2012 Apr 1;56(4):965-72.
50. Lee YA, Wallace MC, Friedman SL. Pathobiology of liver fibrosis: a translational success story. Gut. 2015 May 1;64(5):830-41.
51. Tacke F. Targeting hepatic macrophages to treat liver diseases. Journal of Hepatology. 2017 Jun 1;66(6):1300- 12.
52. Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, et al. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. American Journal of Respiratory and Critical Care Medicine. 2011 Sep 1;184(5):569-81.
53. Li M, Riddle S, Zhang H, D’Alessandro A, Flockton A, Serkova NJ, Hansen KC, et al. Metabolic reprogramming regulates the proliferative and inflammatory phenotype of adventitial fibroblasts in pulmonary hypertension through the transcriptional corepressor C-terminal binding protein-1. Circulation. 2016 Oct 11;134(15):1105-21.
54. El Kasmi KC, Pugliese SC, Riddle SR, Poth JM, Anderson AL, Frid MG, et al. Adventitial fibroblasts induce a distinct proinflammatory/profibrotic macrophage phenotype in pulmonary hypertension. The Journal of Immunology. 2014 Jul 15;193(2):597-609.
55. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. The Journal of Clinical Investigation. 2005 Jan 3;115(1):56-65.
56. Lotowska JM, Sobaniec-Lotowska ME, Lebensztejn DM. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis–ultrastructural findings. The first report in pediatric patients. Scandinavian Journal of Gastroenterology. 2013 Mar 1;48(3):352-7.
57. Friedman JE, Dobrinskikh E, Alfonso-Garcia A, Fast A, Janssen RC, Soderborg TK, et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatology Communications. 2018 Mar;2(3):313-28.
58. Chu DM, Antony KM, Ma J, Prince AL, Showalter L, Moller M, et al. The early infant gut microbiome varies in association with a maternal high-fat diet. Genome Medicine. 2016 Dec;8(1):1-2.
59. Schwartz S, Friedberg I, Ivanov IV, Davidson LA, Goldsby JS, Dahl DB, et al. A metagenomic study of dietdependent interaction between gut microbiota and host in infants reveals differences in immune response. Genome Biology. 2012 Apr 1;13(4):r32.
60. Bokulich NA, Chung J, Battaglia T, Henderson N, Jay M, Li H, et al. Antibiotics, birth mode, and diet shape microbiome maturation during early life. Science Translational Medicine. 2016 Jun 15;8(343):343ra82.
61. Madan JC, Hoen AG, Lundgren SN, Farzan SF, Cottingham KL, Morrison HG, et al. Association of cesarean delivery and formula supplementation with the intestinal microbiome of 6-week-old infants. JAMA Pediatrics. 2016 Mar 1;170(3):212-9.
62. Koch MA, Reiner GL, Lugo KA, Kreuk LS, Stanbery AG, Ansaldo E, et al. Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell. 2016 May 5;165(4):827-41.
63. Mirpuri J, Raetz M, Sturge CR, Wilhelm CL, Benson A, Savani RC, et al. Proteobacteria-specific IgA regulates maturation of the intestinal microbiota. Gut Microbes. 2014 Jan 1;5(1):28-39.
64. Honda K, Littman DR. The microbiota in adaptive immune homeostasis and disease. Nature. 2016 Jul;535(7610):75-84.
65. Hand TW, Vujkovic-Cvijin I, Ridaura VK, Belkaid Y. Linking the microbiota, chronic disease, and the immune system. Trends in Endocrinology & Metabolism. 2016 Dec 1;27(12):831-43.
66. Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Science Translational Medicine. 2015 Sep 30;7(307):307ra152.
67. Nogacka AM, Salazar N, Arboleya S, Suárez M, Fernández N, Solís G, et al. Early microbiota, antibiotics and health. Cellular and Molecular Life Sciences. 2018 Jan 1;75(1):83-91.
68. Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, et al. Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell. 2014 Aug 14;158(4):705-21.
69. Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, et al. Microbial exposure during early life has persistent effects on natural killer T cell function. Science. 2012 Apr 27;336(6080):489-93.
70. Sanidad KZ, Zeng MY. Neonatal gut microbiome and immunity. Current Opinion in Microbiology. 2020 Aug 1;56:30-7.
71. Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nature Reviews Immunology. 2007 May;7(5):379-90.
72. Torow N, Hornef MW. The neonatal window of opportunity: setting the stage for life-long host-microbial interaction and immune homeostasis. The Journal of Immunology. 2017 Jan 15;198(2):557-63.
73. Fulde M, Hornef MW. Maturation of the enteric mucosal innate immune system during the postnatal period. Immunological Reviews. 2014 Jul;260(1):21-34.
74. Rakoff-Nahoum S, Kong Y, Kleinstein SH, Subramanian S, Ahern PP, Gordon JI, et al. Analysis of gene–environment interactions in postnatal development of the mammalian intestine. Proceedings of the National Academy of Sciences. 2015 Feb 17;112(7):1929-36.
75. Kabat AM, Srinivasan N, Maloy KJ. Modulation of immune development and function by intestinal microbiota. Trends in Immunology. 2014 Nov 1;35(11):507- 17.
76. Funkhouser LJ, Bordenstein SR. Mom knows best: the universality of maternal microbial transmission. PLoS Biology. 2013 Aug 20;11(8):e1001631.
77. McCoy KD, Thomson CA. The impact of maternal microbes and microbial colonization in early life on hematopoiesis. The Journal of Immunology. 2018 Apr 15;200(8):2519-26.
78. de Agüero MG, Ganal-Vonarburg SC, Fuhrer T, Rupp S, Uchimura Y, Li H, et al. The maternal microbiota drives early postnatal innate immune development. Science. 2016 Mar 18;351(6279):1296-302.
79. Negi S, Das DK, Pahari S, Nadeem S, Agrewala JN. Potential role of gut microbiota in induction and regulation of innate immune memory. Frontiers in Immunology. 2019;10:2441.
80. Gorjifard S, Goldszmid RS. Microbiota—myeloid cell crosstalk beyond the gut. Journal of Leukocyte Biology. 2016 Nov;100(5):865-79.
81. Weaver LK, Minichino D, Biswas C, Chu N, Lee JJ, Bittinger K, Albeituni S, Nichols KE, Behrens EM. Microbiota-dependent signals are required to sustain TLR-mediated immune responses. JCI Insight. 2019 Jan 10;4(1):e124370.
82. McCoy KD, Burkhard R, Geuking MB. The microbiome and immune memory formation. Immunology and Cell Biology. 2019 Aug;97(7):625-35.
83. Mitroulis I, Ruppova K, Wang B, Chen LS, Grzybek M, Grinenko T, et al. Modulation of myelopoiesis progenitors is an integral component of trained immunity. Cell. 2018 Jan 11;172(1-2):147-61.
84. Kleinnijenhuis J, Quintin J, Preijers F, Joosten LA, Ifrim DC, Saeed S, et al. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proceedings of the National Academy of Sciences. 2012 Oct 23;109(43):17537-42.
85. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. 2016 Apr 22;352(6284):aaf1098.
86. Vatanen T, Kostic AD, d’Hennezel E, Siljander H, Franzosa EA, Yassour M, et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell. 2016 May 5;165(4):842-53.
87. Thorburn AN, McKenzie CI, Shen SJ, Stanley D, Macia L, Mason LJ, et al. Evidence that asthma is a developmental origin disease influenced by maternal diet and bacterial metabolites. Nature Communications. 2015 Jun 23;6(1):1-3.
88. Nakajima A, Kaga N, Nakanishi Y, Ohno H, Miyamoto J, Kimura I, et al. Maternal high fiber diet during pregnancy and lactation influences regulatory T cell differentiation in offspring in mice. The Journal of Immunology. 2017 Nov 15;199(10):3516-24.
89. Levy M, Thaiss CA, Elinav E. Metabolites: messengers between the microbiota and the immune system. Genes & Development. 2016 Jul 15;30(14):1589-97.
90. Laforest-Lapointe I, Arrieta MC. Patterns of earlylife gut microbial colonization during human immune development: an ecological perspective. Frontiers in Immunology. 2017 Jul 10;8:788.
91. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. 2007 Jun 26;5(7):e177.
92. Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, et al. Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics. 2006 Aug 1;118(2):511-21.
93. Mitsuoka T. Intestinal ?ora and human health. Asia Pacific Journal of Clinical Nutrition. 1996;5:2-9.
94. Ifrim DC, Quintin J, Joosten LA, Jacobs C, Jansen T, Jacobs L, et al. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clinical and Vaccine Immunology. 2014 Apr 1;21(4):534- 45.
95. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends in Immunology. 2009 Oct 1;30(10):475-87.
96. Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. Journal of Endotoxin Research. 2004 Apr;10(2):71-84.
97. Hofstad T, Sveen K, Dahlen G. Chemical composition, serological reactivity and endotoxicity of lipopolysaccharides extracted in different ways from Bacteroides fragilis, Bacteroides melaninogenicus and Bacteroides oralis. Acta Pathologica Microbiologica Scandinavica Section B Microbiology. 1977 Sep;85(4):262- 70.
98. Lemas DJ, Young BE, Baker PR, Tomczik AC, Soderborg TK, Hernandez TL, et al. Alterations in human milk leptin and insulin are associated with early changes in the infant intestinal microbiome. The American Journal of Clinical Nutrition. 2016 May 1;103(5):1291-300.
99. Soderborg TK, Clark SE, Mulligan CE, Janssen RC, Babcock L, Ir D, et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nature Communications. 2018 Oct 26;9(1):1-2.
100. Schwimmer JB, Behling C, Newbury R, Deutsch R, Nievergelt C, Schork NJ, et al. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology. 2005 Sep;42(3):641-9.
101. Brunt EM, Kleiner DE, Wilson LA, Unalp A, Behling CE, Lavine JE, et al. Portal chronic inflammation in nonalcoholic fatty liver disease (NAFLD): a histologic marker of advanced NAFLD—clinicopathologic correlations from the nonalcoholic steatohepatitis clinical research network. Hepatology. 2009 Mar;49(3):809-20.
102. Perdiguero EG, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015 Feb;518(7540):547-51.
103. Hoeffel G, Ginhoux F. Fetal monocytes and the origins of tissue-resident macrophages. Cellular Immunology. 2018 Aug 1;330:5-15.
104. Mikkola HK, Orkin SH. The journey of developing hematopoietic stem cells. Development. 2006 Oct 1;133(19):3733-44.
105. Sheng J, Ruedl C, Karjalainen K. Most tissueresident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. 2015 Aug 18;43(2):382-93.
106. Pittet MJ, Nahrendorf M, Swirski FK. The journey from stem cell to macrophage. Annals of the New York Academy of Sciences. 2014 Jun;1319(1):1.
107. Luo Y, Chen GL, Hannemann N, Ipseiz N, Krönke G, Bäuerle T, et al. Microbiota from obese mice regulate hematopoietic stem cell differentiation by altering the bone niche. Cell Metabolism. 2015 Nov 3;22(5):886-94.
108. Broxmeyer HE, Hangoc G, Cooper S, Ribeiro RC, Graves V, Yoder M, et al. Growth characteristics and expansion of human umbilical cord blood and estimation of its potential for transplantation in adults. Proceedings of the National Academy of Sciences. 1992 May 1;89(9):4109- 13.
109. Gourbal B, Pinaud S, Beckers GJ, Van Der Meer JW, Conrath U, Netea MG. Innate immune memory: an evolutionary perspective. Immunological Reviews. 2018 May;283(1):21-40.
110. Hergott CB, Roche AM, Tamashiro E, Clarke TB, Bailey AG, Laughlin A, et al. Peptidoglycan from the gut microbiota governs the lifespan of circulating phagocytes at homeostasis. Blood. 2016 May 19;127(20):2460-71.
111. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG educates hematopoietic stem cells to generate protective innate immunity against tuberculosis. Cell. 2018 Jan 11;172(1-2):176-90.
112. Kleinnijenhuis J, Quintin J, Preijers F, Benn CS, Joosten LA, Jacobs C, et al. Long-lasting effects of BCG vaccination on both heterologous Th1/Th17 responses and innate trained immunity. Journal of Innate Immunity. 2014;6(2):152-8.
113. Quintin J, Saeed S, Martens JH, Giamarellos- Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LA. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host & Microbe. 2012 Aug 16;12(2):223-32.
114. Kamimae-Lanning AN, Krasnow SM, Goloviznina NA, Zhu X, Roth-Carter QR, Levasseur PR, et al. Maternal high-fat diet and obesity compromise fetal hematopoiesis. Molecular Metabolism. 2015 Jan 1;4(1):25-38.
115. Singer K, DelProposto J, Morris DL, Zamarron B, Mergian T, Maley N, et al. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Molecular Metabolism. 2014 Sep 1;3(6):664-75.
116. Christ A, Günther P, Lauterbach MA, Duewell P, Biswas D, Pelka K, et al. Western diet triggers NLRP3- dependent innate immune reprogramming. Cell. 2018 Jan 11;172(1-2):162-75.
117. Friedman JE, Dobrinskikh E, Alfonso-Garcia A, Fast A, Janssen RC, Soderborg TK, et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatology Communications. 2018 Mar;2(3):313-28.
118. Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR-and HIF-1a–mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014 Sep 26;345(6204):1250684.
119. Mulder WJ, Ochando J, Joosten LA, Fayad ZA, Netea MG. Therapeutic targeting of trained immunity. Nature Reviews Drug Discovery. 2019 Jul;18(7):553-66.
120. van der Meer JW, Joosten LA, Riksen N, Netea MG. Trained immunity: a smart way to enhance innate immune defence. Molecular Immunology. 2015 Nov 1;68(1):40-4.
121. Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, et al. Interferon gamma induces reversible metabolic reprogramming of M1 macrophages to sustain cell viability and pro-inflammatory activity. EBioMedicine. 2018 Apr 1;30:303-16.
122. McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature. 2013;496:238242.
123. Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Research. 2015 Jul;25(7):771-84.
124. Benit P, Letouze E, Rak M, Aubry L, Burnichon N, Favier J, et al. Unsuspected task for an old team: succinate, fumarate and other Krebs cycle acids in metabolic remodeling. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2014 Aug 1;1837(8):1330-7.
125. Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, et al. a-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nature Immunology. 2017 Sep;18(9):985-94.
126. Baardman J, Licht I, De Winther MP, Van den Bossche J. Metabolic–epigenetic crosstalk in macrophage activation. Epigenomics. 2015 Oct;7(7):1155-64.
127. Wang X, Cao Q, Yu L, Shi H, Xue B, Shi H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight. 2016 Nov 17;1(19):87748.