Abstract
We reviewed the anti-tumor mechanisms of short-chain fatty acids (SCFAs) as well as the relationship between the gut microbiome and the pathology of colorectal carcinoma (CRC). According to our in silico analysis of human CRC cell lines, it was shown that SCFAs suppress various genes and transcription factors that participate in tumor growth/proliferation and cell turnover, and butyric acid displayed the strongest inhibitory effects among SCFAs. Metagenomics analysis of fecal and tumor epithelial microbiomes in patients with CRC suggested that the disease progression of CRC is affected by the crosstalk between carcinoma epithelial cells and gut microbes. No microbes producing SCFAs were identified in the fecal or tumor epithelial microbiomes of patients with CRC. Our findings also suggested that microbes in the feces and tumor epithelia of patients with CRC do not affect oncogenes or carcinogenic pathway.
Keywords
Gut microbiome, Colorectal carcinoma, Short-chain fatty acids (SCFAs), Colonic epithelium-microbiome crosstalk, Tumor growth and proliferation, In silico analysis
Abbreviations
CRC: Colorectal Carcinoma; IPA: Ingenuity Pathway Analysis; MA_FC: Microarray Fold-Change; SCFA: Short-Chain Fatty Acid
Introduction
Short-chain fatty acids (SCFAs) produced by the gut microbiome have been reported to have anti-tumor effects in several experimental systems [1-3]. Previously, we investigated the inhibitory effects of SCFAs (butyric acid, isobutyric acid, and acetic acid) on cell growth and proliferation in cultured human colorectal carcinoma (CRC) cell lines (DLD-1 cells, WirDr cells), and found that butyric acid displayed the strongest inhibitory effect [4]; however, the underlying mechanisms have not yet been elucidated. To investigate the anti-tumor mechanisms of SCFAs, we performed an in silico analysis of their inhibitory mechanism on tumor cell growth and proliferation in an experimental system in which SCFAs were added to cultured human CRC cell lines [5]; the results revealed that SCFAs suppress genes and transcription factors that participate in tumor cell growth, proliferation, and turnover, but do not affect genes involved in carcinogenesis, or genomes and factors associated with carcinogenic pathways.
This motivated our subsequent investigation of the gut microbiomes of patients with CRC [6], including the functions and roles of the identified microbial species in colorectal carcinogenesis and tumor growth and proliferation. Metagenomics analysis of the feces and tumor epithelial cells of patients with CRC identified common microorganisms in tumor epithelia of CRC and elucidated their various functions [6].
In the present review, we describe the relationship between the pathophysiology of CRC and the gut microbiome of patients with CRC, including a discussion of the current knowledge base and major focus areas for future research.
Inhibitory Actions of SCFAs in Human CRC Cell Lines
Although SCFAs have been reported to display anti-tumor activities, it is known that these activities differ depending on the dose. In a previous study [4], human CRC cell lines (DLD- 1 cells, WirDr cells) were cultured with three different SCFAs (butyric acid, isobutyric acid, and acetic acid) at 11 different doses to compare their in vitro anti-tumor effects. The cell growth inhibitory activity was quantified using a commercial WST-8 assay, based on the measurement of spectrophotometric absorbance (450 nm) of water-soluble formazan formed via reduction by intracellular dehydrogenase. The IC50 value, representing the concentration of SCFA that achieved 50% inhibition of cell growth, was calculated by plotting the cell count of the control culture against the SCFA concentration. The IC50 values indicated that the three SCFAs displayed tumor growth-inhibitory actions in the descending order of butyric acid, isobutyric acid, and acetic acid (Table 1).
| IC50 (nM, mean ± SEM, n=4) | |||
|---|---|---|---|
| Butyric acid | Isobutyric acid | Acetic acid | |
| DLD-1 cell | 2.89 ± 0.29 | 12.9 ± 0.80 | 16.6 ± 1.10 |
| WirDr cell | 1.59 ± 0.04 | 10.2 ± 0.77 | 15.1 ± 1.25 |
Table 1: Clinical trials of natural and synthetic mTOR inhibitors against COVID-19.
Inhibitory Mechanisms of SCFAs against Tumor Cell Growth and Proliferation
An in silico analysis, allows one-time, comprehensive analyses of biological functions, genes, molecular networks, and biological pathways in a living body, based on various input data and tools that form part of the bioinformatics approach, such as microarrays, RNA sequencing, metabolomics, and proteomics. In particular, the Kyoto Encyclopedia of Genes and Genomes and Ingenuity Pathway Analysis (IPA) are commonly used as software platforms for in silico analyses [7,8]. An in silico approach to genomics research is considered an epoch-making innovation as it allows for time- and costefficient analyses using a personal computer. This analysis will be upgraded in a timely manner. It is a versatile approach that enables visualization of various numerical data, including data from tumor pathophysiological analyses [9-11], in addition to allowing untargeted analysis of large data sets and comprehensive analysis of the interrelationship between pathophysiological mechanisms. However, it is essential to evaluate and confirm the validity of the results of in silico analyses using knowledge obtained from wet-lab experiments in vitro and in vivo.
We analyzed the anti-tumor mechanisms of SCFAs in DLD- 1 cells using an in silico approach [5]. DNA microarrays were used to identify groups of differentially expressed genes by determining and adjusting the total RNA concentration of cells treated with acetic acid, butyric acid, and isobutyric acid at their IC50 values (Table 1) for 24 h. Gene groups with expression levels as 50% of those of untreated cells were subjected to IPA analysis. Transcriptome Analysis Console software (version 3.1; Applied Biosystems, Waltham, MA, USA) was used to analyze the gene expression data. Genes were filtered using microarray fold-change (MA_FC) values. The genes that scored MA_FC < 5 were subsequently analyzed using IPA software to identify canonical pathways, biological functions, gene networks, and key processes that were enriched and/or modulated effectively by SCFAs in DLD-1 cells. The raw microarray data files obtained from these analyses were presented to the Gene Expression Omnibus.
In the gene group whose expression level was increased by 50% or more (shown in blue), there was no significant change in gene functions based on the results of IPA analyses [5]. On the contrary, the change in gene functions was remarkable in the gene group (shown in green) whose expression level was reduced to 50% or less. We analyzed the effects on the cell cycle, proliferation, and DNA replication function of genes whose expression levels were reduced to 50% or less after 24-h treatment with butyric acid in DLD-1 cells (Figure 1A), and performed an anti-tumor pathway analysis, as well as heat map analyses of disease and function. Similar analyses were performed for isobutyric acid and acetic acid (Figures 1B and 1C). A total of 791 genes, including E2F1, UHRF1, HIST2H3A, HIST1H4K, HIST1H4L, HIST1H3B, HIST1H3D, HIST1H3H, and FOXM1, participated in the cell cycle, cell proliferation, and DNA replication of almost all CRC cells under investigation. Furthermore, the results of pathway and heatmap analyses of butyric acid (Figures 2A and 3A), isobutyric acid (Figures 2B and 3B), and acetic acid (Figures 2C and 3C) indicated that butyric acid had the strongest anti-tumor action among the three SCFAs [5]. Notably, it was found that the SCFAs scarcely suppressed the expression of cancer-related genes, but strongly suppressed tumor metabolic molecules (Figure 3), including tumor cell cycle, DNA replication, recombination, and repair.
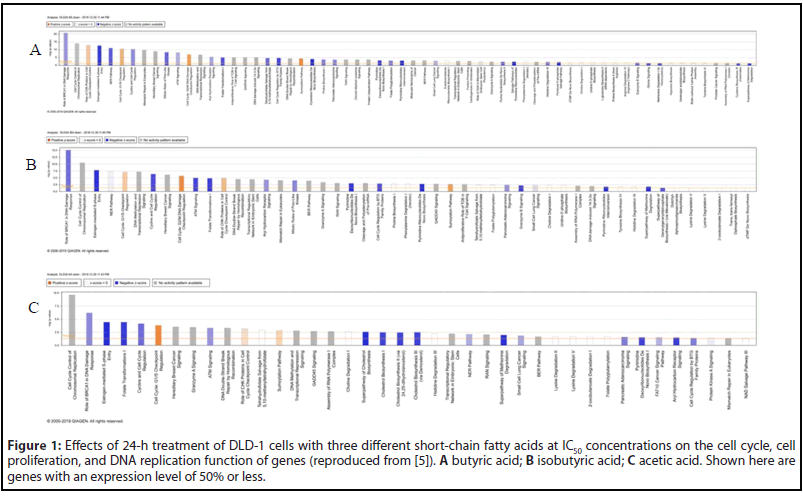
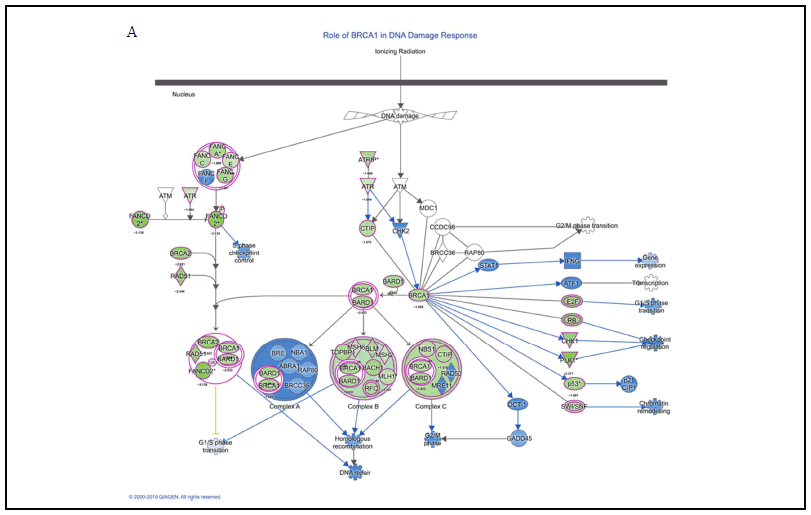
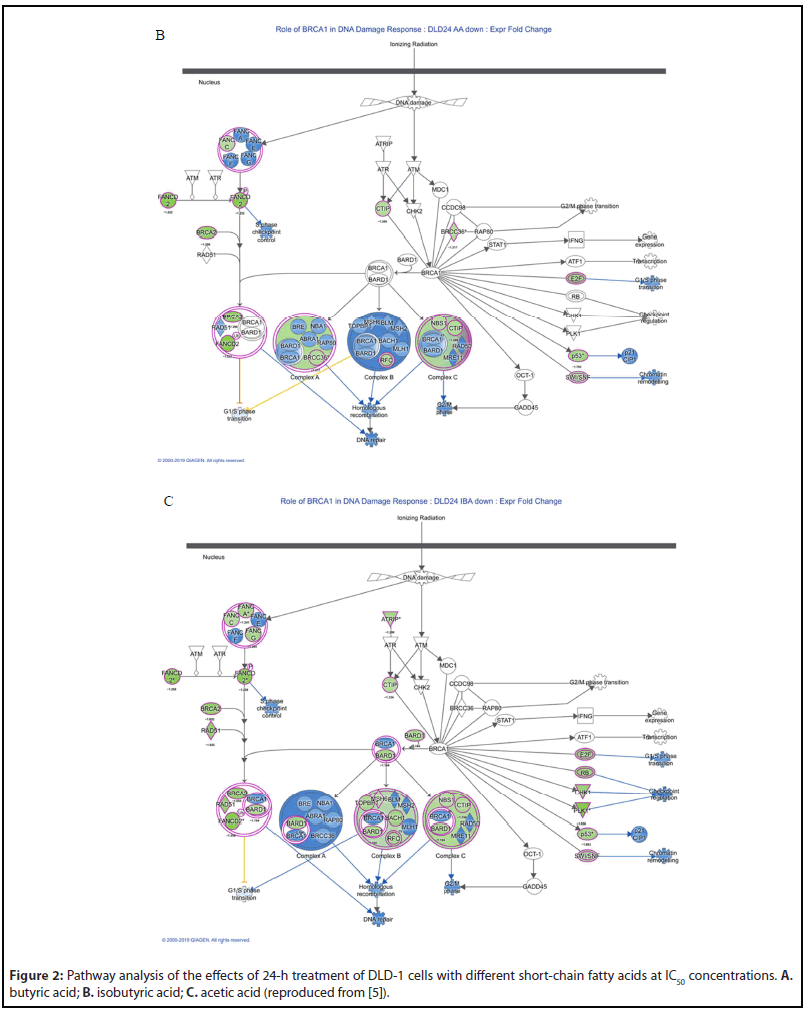
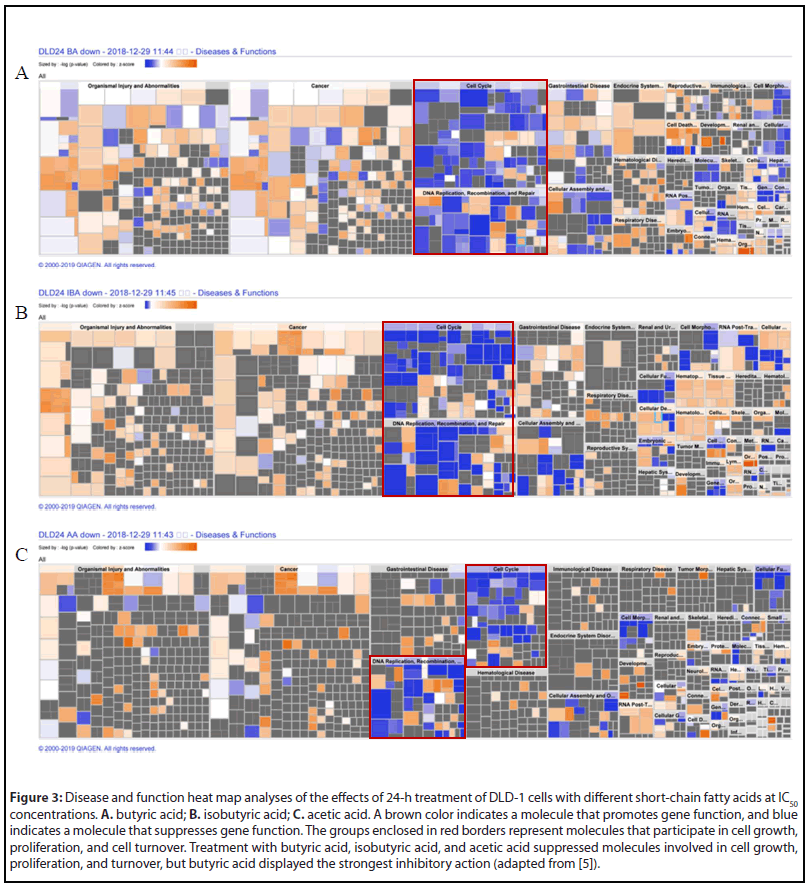
Overall, these finding contributed substantially to our general understanding of the anti-tumor mechanism of SCFAs, and represented the first comprehensive report on the anti-colorectal tumor mechanism of SCFAs, pointing the way toward future studies on CRC prevention research. It has been reported that butyric acid is produced not only by gut microbes but also in colonic mucin layers [12,13]. Therefore, future studies should investigate the relationship between disease progression in patients with CRC and SCFAs produced by the gut microbiome, colonic mucus layers, and exosomes secreted by gut microbes.
Relationship between the Gut Microbiome and CRC
The relationship between the gut microbiome and cell proliferation in colorectal carcinogenesis was explored further. We obtained fecal samples and tumor epithelial cells from eighteen patients with advanced CRC located at various sites within the colorectum [6], and extracted genomic DNA from each sample for metagenomics analysis. Heat map clustering of microbial species identified in different colorectal areas was performed using 16S rRNA amplicon sequences (Figure 4A), while the Quantitative Insights Into Microbial Ecology (QIIME) and Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) bioinformatics software packages were used for structural and functional analyses of the microbiomes, respectively [6]. The structural analysis of fecal microbes showed differences depending on the tumor site (Figure 4B), but the pathophysiological functional analysis of microbes revealed near-uniform functional similarities regardless of the tumor site (Figure 4C).
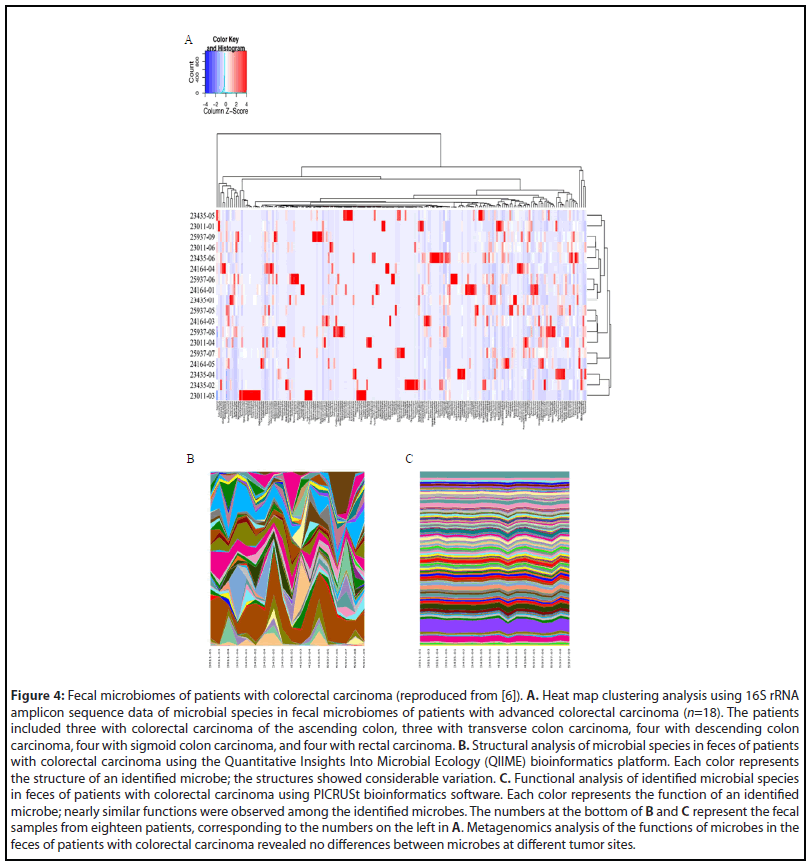
Among the eighteen patients with CRC, three with descending colon carcinoma, three with sigmoid colon carcinoma, and three with rectal carcinoma were selected for the identification of common microorganisms, including analyses of their structures and functions [6]. We performed a heat map clustering analysis, as well as QIIME and PICRUSt analyses (Figures 5B and 5C, respectively), of the microbial species in the fecal microbiomes of patients with advanced colorectal carcinoma in different parts of the colorectum. The functional analysis using PICRUSt identified genes involved in several functions and systems: amino acid and ATP-binding cassette transporters; phosphotransferase system; chaperone system; DNA repair, recombination, and replication; transcription factor; ribosome; cysteine, methionine, phenylalanine, tyrosine, valine, leucine, isoleucine, alanine, aspartic acid, and glutamine metabolism; tryptophan affects biosynthesis and metabolism; amino acidrelated enzymes; tricarboxylic acid (TCA) circuit; fructose and mannose metabolism; glycolysis and glycation; purine and pyrimidine metabolism; pentose phosphate pathway; energy metabolism; nitrogen metabolism; methane metabolism; and one carbon pool by folic acid. All of these are essential molecules or factors that participate in tumor growth and proliferation.
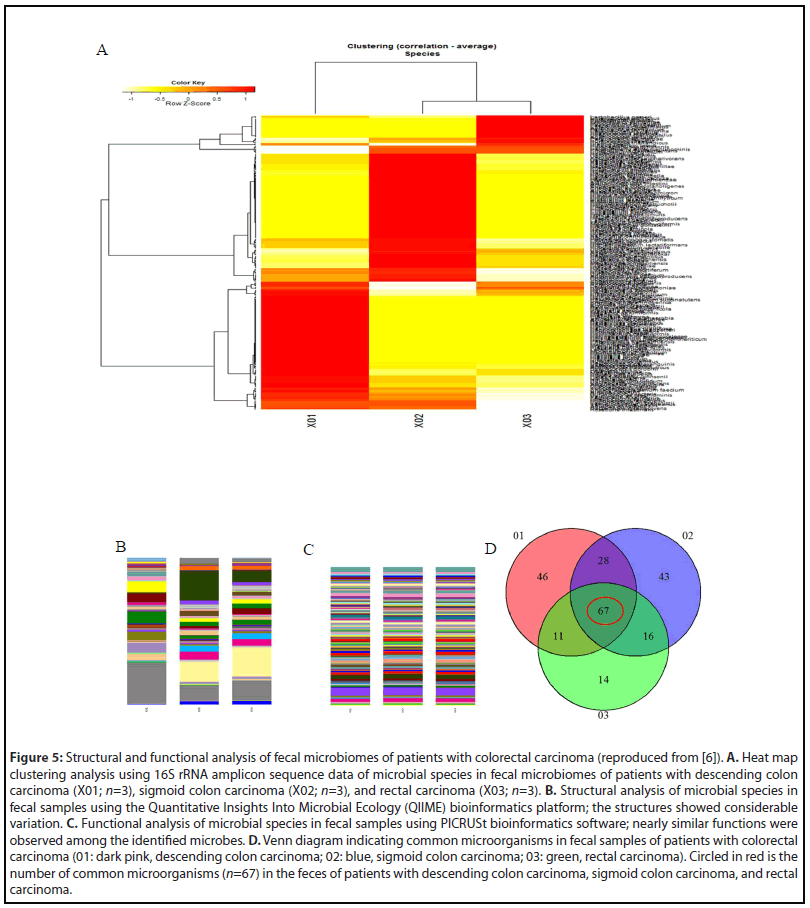
The Venn diagram is an analytical method for identifying mutual microbes in different groups. Sixty-seven microbial species were identified by Venn diagram as common microorganisms in the feces of patients with CRC (Figure 5D, Table 2).
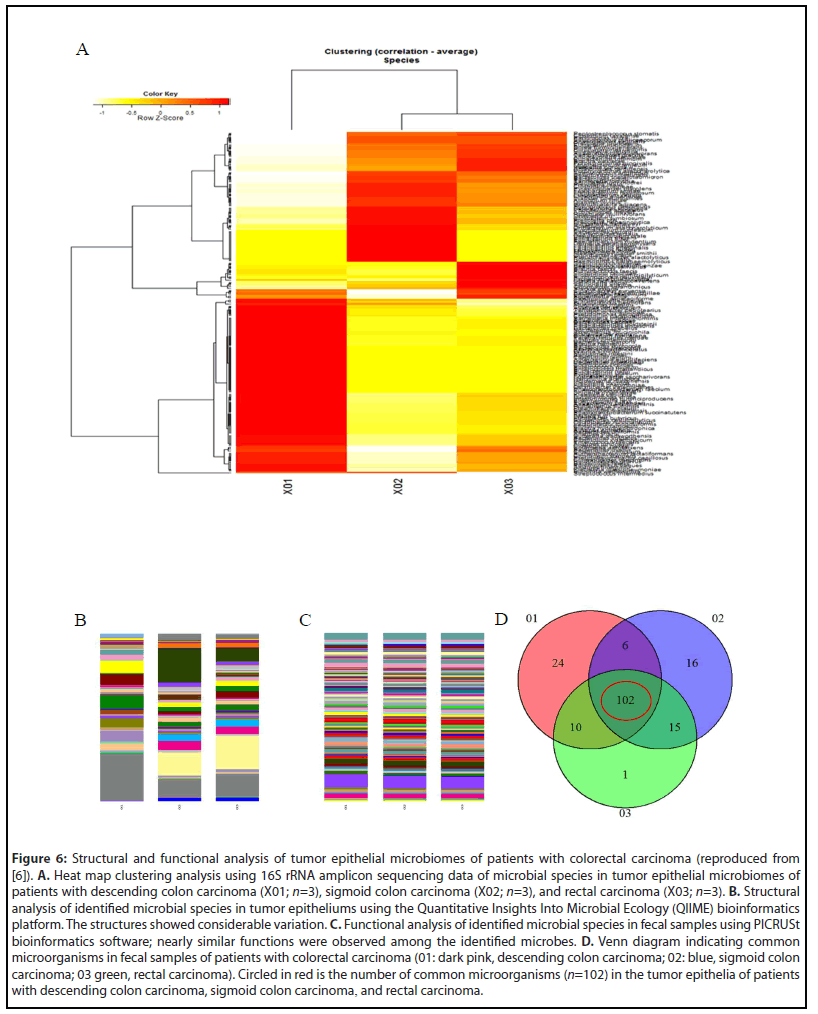
An evaluation of the microbiomes in tumor epithelia of the aforementioned patients using heat map clustering data (Figure 6A) indicated that the microbial species differed from those observed in the feces of the same patients [6]. However, the QIIME analysis of molecular structures (Figure 6B) showed results similar to those observed in the fecal samples. Moreover, the identified microbes in the tumor epithelia had functions similar to those of fecal microbes, based on the PICRUSt analysis (Figure 6C). These functions include: amino acid transporters; ABC transporters; amino acid-related enzymes; leucine, valine, isoleucine, methionine, phenylalanine, and tryptophan biosynthesis and metabolism; glycolysis and gluconeogenesis; TCA cycle; pentose phosphate pathway; and pyruvate metabolism. Furthermore, these microbes also had functions that affect purine/pyrimidine metabolism, DNA repair/replication, protein exports, RNA degradation, and transcription factors. Among the patients that were studied, a total of 102 common microorganisms were identified in the tumor epithelia (Figure 6D, Table 3) [6].

According to the functional analysis of mutual microbes in fecal and tumor epithelial microbiomes of patients with CRC, there were no microbes that activate any carcinogenic pathways or cancer-related signals. The identified microbes were shown to participate in tumor metabolism and tumor growth, including expression of transporters essential for amino acid uptake, DNA replication and repair, RNA degradation, and modulation of transcription factors [6]. Overall, the study suggests that CRC grows and proliferates due to crosstalk between the gut microbiota and carcinoma epithelial cells. However, the mechanism of colorectal carcinogenesis remains unknown.
We have described that SCFAs suppress factors that participate in the growth and proliferation of colorectal tumors. Faecalibacterium prausnitzii and Roseburia hominis in Clostridium cluster IV, and Clostridium butyricum and Eubacterium rectale included in Clostridium cluster XIVa, common microorganisms in the feces and tumor epithelia of patients with CRC, were not identified. These are representative examples of SCFA-producing microbes.
Currently, the relationship between CRC and Fusobacterium nucleatum is drawing attention, as this microbe has been implicated in colorectal carcinogenesis [14-16]. In particular, it has been noted that F. nucleatum, a common microorganism in tumor epithelia of patients with CRC (Table 3) [6], has the ability to avoid the immune system involved in carcinogenesis [17-19]. Recent reports suggest that F. nucleatum participates in tumor growth, proliferation, and metastasis rather than in carcinogenesis [20-22]. It has been reported that high counts of F. nucleatum are detected in the feces of patients with CRC. Consequently, a method for screening for CRC by detecting F. nucleatum in the feces of CRC has been reported [23,24].
To clarify the relationship between CRC and the gut microbiome, large patient cohorts are required to obtain robust data, identify common microorganisms CRC, and analyze their functions in detail. Furthermore, it is necessary to verify the analysis by performing shotgun sequencing or long-read sequencing in addition to 16S rRNA amplicon sequencing.
Conclusions
We discussed the connections between CRC, the gut microbiome, anti-colorectal tumor effects of SCFAs, and the underlying mechanisms. Although the mechanisms of colorectal carcinogenesis are unknown, it has been suggested that CRC grows and proliferates through the crosstalk between the gut microbiome and cancer epithelial cells. Metagenomic analysis suggested that common microorganisms CRC did not influence oncogenes or routes related to carcinogenesis. Furthermore, the available data suggest that SCFAs produced by gut microflora also exert anti-colorectal tumor effects by suppressing pathways associated with tumor growth, proliferation, and cell turnover from an in silico analysis method. However, no common microorganism that are associated with the production of SCFAs were identified in the fecal and tumor epithelial microbiomes of CRC.
Future research should include large numbers of cases and analyze the production of SCFAs by the gut microbes as well as the colonic mucus layers. Furthermore, a comprehensive analysis of the relationship between tumor-infiltrating T cell repertoires and nucleic acids in exosomes secreted by microbes is also required. Finally, the relationship of CRC with the gut virome, including phages, has recently attracted attention, and requires further investigation.
Author Contributions
Tadashi Ohara was in charge of the culture experiments, DNA microarray part, IPA analysis and microbial data analysis. Yasuyuki Taki supported this research as a whole.
Conflict of Interest
There is no conflict of interest.
Acknowledgements
No company has been funded for this study. Tsutomu Mori Ph.D. of the Department of Human Lifesciences, School of Nursing, Fukushima Medical University supported of all the data mining and IPA analysis of the results. The analysis of microbiota was carried out with the cooperation of Techno Suruga Lab (Shizuoka, Japan). The author would like to thank all those who have cooperated in this research.
References
2. Yuan X, Xue J, Tan Y, Yang Q, Qin Z, Bao X, et al. Albuca Bracteate Polysaccharides Synergistically Enhance the Anti-Tumor Efficacy of 5-Fluorouracil Against Colorectal Cancer by Modulating ß-Catenin Signaling and Intestinal Flora. Frontiers in Pharmacology. 2021:736627.
3. Li B, Liu M, Wang Y, Gong S, Yao W, Li W, et al. Puerarin improves the bone micro-environment to inhibit OVX-induced osteoporosis via modulating SCFAs released by the gut microbiota and repairing intestinal mucosal integrity. Biomedicine & Pharmacotherapy. 2020 Dec 1;132:110923.
4. Ohara T, Suzutani T. Intake of Bifidobacterium longum and Fructooligosaccharides prevents Colorectal Carcinogenesis. Euroasian Journal of Hepato-gastroenterology. 2018 Jan;8(1):11-17.
5. Ohara T, Mori T. Antiproliferative effects of short-chain fatty acids on human colorectal cancer cells via gene expression inhibition. Anticancer Research. 2019 Sep 1;39(9):4659-66.
6. Ohara T. Colorectal Cancer Grows and Proliferates Due to Crosstalk Between Intestinal Microbiota and Mucosal Epithelium. Cancer Sci Res. 2021;4(4):1-2.
7. Calderón-González KG, Hernández-Monge J, Herrera-Aguirre ME, Luna-Arias JP. Bioinformatics tools for proteomics data interpretation. Modern Proteomics–Sample Preparation, Analysis and Practical Applications. 2016:281-341.
8. Wang F, Lu J, Peng X, Wang J, Liu X, Chen X, et al. Integrated analysis of microRNA regulatory network in nasopharyngeal carcinoma with deep sequencing. Journal of Experimental & Clinical Cancer Research. 2016 Dec;35(1):17.
9. Wang G, Dong F, Xu Z, Sharma S, Hu X, Chen D, et al. MicroRNA profile in HBV-induced infection and hepatocellular carcinoma. BMC Cancer. 2017 Dec;17(1):805.
10. Pang K, Hao L, Shi Z, Chen B, Pang H, Dong Y, et al. Comprehensive gene expression analysis after ERH gene knockdown in human bladder cancer T24 cell lines. Gene. 2020 May 15;738:144475.
11. Chen X, Liao L, Li Y, Huang H, Huang Q, Deng S. Screening and Functional Prediction of Key Candidate Genes in Hepatitis B Virus-Associated Hepatocellular Carcinoma. BioMed Research International. 2020 Oct 9;2020:7653506.
12. Van den Abbeele P, Belzer C, Goossens M, Kleerebezem M, De Vos WM, Thas O, et al. Butyrate-producing Clostridium cluster XIVa species specifically colonize mucins in an in vitro gut model. The ISME Journal. 2013 May;7(5):949-61.
13. Yamada T, Hino S, Iijima H, Genda T, Aoki R, Nagata R, et al. Mucin O-glycans facilitate symbiosynthesis to maintain gut immune homeostasis. EBioMedicine. 2019 Oct 1;48:513-25.
14. Hashemi Goradel N, Heidarzadeh S, Jahangiri S, Farhood B, Mortezaee K, Khanlarkhani N, et al. Fusobacterium nucleatum and colorectal cancer: A mechanistic overview. Journal of Cellular Physiology. 2019 Mar;234(3):2337-44.
15. Hong J, Guo F, Lu SY, Shen C, Ma D, Zhang X, et al. F. nucleatum targets lncRNA ENO1-IT1 to promote glycolysis and oncogenesis in colorectal cancer. Gut. 2021 Nov 1;70(11):2123-37.
16. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/ß-catenin signaling via its FadA adhesin. Cell Host & Microbe. 2013 Aug 14;14(2):195-206.
17. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host & Microbe. 2013 Aug 14;14(2):207-15.
18. Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncology. 2015 Aug 1;1(5):653-61.
19. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity. 2015 Feb 17;42(2):344-55.
20. Ohara T. Colorectal carcinogenesis, colorectal cancer development and Fusobacterium nucleatum. Front Drug Chem Clin Res. 2021; 4:1-4.
21. Amitay EL, Werner S, Vital M, Pieper DH, Höfler D, Gierse IJ, et al. Fusobacterium and colorectal cancer: causal factor or passenger? Results from a large colorectal cancer screening study. Carcinogenesis. 2017 Aug 1;38(8):781-8.
22. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nature Medicine. 2019 Jun;25(6):968-76.
23. Zhang X, Zhu X, Cao Y, Fang JY, Hong J, Chen H. Fecal Fusobacterium nucleatum for the diagnosis of colorectal tumor: A systematic review and meta-analysis. Cancer Medicine. 2019 Feb;8(2):480-91.
24. Grobbee EJ, Lam SY, Fuhler GM, Blakaj B, Konstantinov SR, Bruno MJ, et al. First steps towards combining faecal immunochemical testing with the gut microbiome in colorectal cancer screening. United European Gastroenterology Journal. 2020 Apr;8(3):293- 302.