Abstract
Hyperglycemia in diabetes induces impairment of hematopoiesis, an important consequence in bone marrow (BM) that contributes to chronic complications in advanced diabetes. The alterations to blood cells associated with diabetes mellitus (DM) pathologies have been carefully and extensively documented, but the underlying mechanism(s) is still unclear. Our recent publication indicates that aberrant intracellular synthesis of hyaluronan (HA) by hyperglycemic dividing BM progenitors is the central mechanism involved. This study demonstrated that macrophages that divided from progenitor cells in hyperglycemia are pro-inflammatory (Mpi) and that the presence of low concentrations of heparin (~20 nM) prevented the intracellular HA synthesis and promoted a tissue repair (Mtr) phenotype. Here, we briefly describe how our new studies of abnormal intracellular hyaluronan synthesis impairs hematopoiesis in diabetes and its regulation by heparin.
Keywords
Diabetes, Hematopoiesis, Inflammation, Monocyte, Macrophage, Hyaluronan, Heparin, Hyperglycemia, Monocyte adhesive hyaluronan matrix
Introduction
Diabetes, a disease that features hyperglycemia, is a major health epidemic at this time. It affects ~34 million people in the US (10.5% of the population) according to the 2020 National Diabetes Statistics Report from the Centers for Disease Control and Prevention (CDC). Nearly 34.5% of the adult population is hyperglycemic enough to be considered pre-diabetic [1]. Prolonged highly elevated blood glucose levels, hyperglycemia, create metabolic stresses that are associated with a wide variety of serious complications involving chronic inflammation in multiple organ systems, including renal disease, cardiovascular and peripheral vascular diseases, peripheral neuropathy that delays wound healing, chronic infections to feet and legs, and ulcerative colitis [1-4]. Further, chronic elevated blood glucose has also been linked to heart attacks and death in subjects with no diagnosis of diabetes mellitus (DM), coronary heart disease, or history of heart failure [3]. However, in almost all cases the relationship of elevated glucose levels to underlying disease mechanisms has not been fully defined, but these diabetic complications do share a common association with extensive and chronic inflammation due to infiltration by activated leukocytes that originate from the bone marrow (BM).
Hyaluronan Synthesis and Its Regulation by Hyperglycemia and Heparin
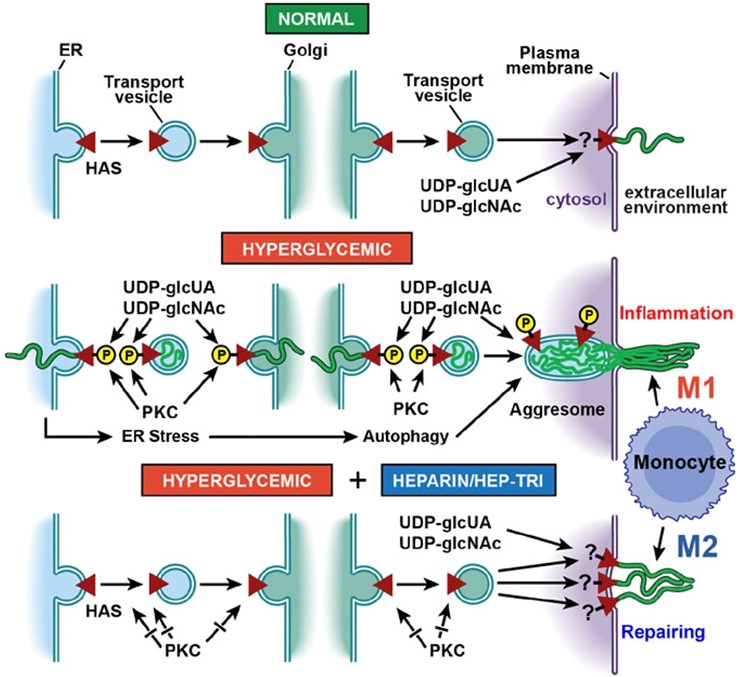
Hyaluronan (HA) is a fascinating macromolecule with a simple structure and a very complex biology. HA is a unique member of the glycosaminoglycan (GAG) family, which includes heparin and chondroitin sulfate (CS). Structurally, HA is the simplest GAG, consisting of a disaccharide, N-acetyl-glucosamine-glucuronic acid (GlcNAc-GlcUA) with one negative charge, that can be repeated >10,000 times (MW 5-10 MDa) due to its unique synthesis. The hydrophilic viscous properties of HA allow it to occupy large hydrodynamic volumes. Further, by interaction with many HA-binding proteins, HA also has a central role in many normal and disease processes that involve cellular signaling and inflammation [5]. HA is synthesized in non-dividing cells by hyaluronan synthases (HASs) that are transported from the endoplasmic reticulum (ER) to the plasma membrane in an inactive form (Figure 1, Normal). Once embedded in the plasma membrane they are activated and alternately add cytosolic UDP-GlcNAc and UDP-GlcUA and extrude the growing GlcNAc-GlcUA disaccharide chains through the plasma membrane to form the cell surface coats and extracellular HA matrices [5-7] that have important physiological roles in maintaining the normal cellular and tissue functions.
Under hyperglycemia, the increased influx of glucose into the dividing cells initiates a protein kinase C (PKC) response that activates hyaluronan synthase (HAS) in intracellular compartments. The glucose stress elevates the cytosolic substrates (UDP-GlcUA and UDP-GlcNAc), which is inhibited by initiating intracellular synthesis of HA, which deposits this high MW (5-10 MDa), polyanionic glycosaminoglycan in intracellular compartments (endolasmic reticulum (ER), transport vesicles, golgi) [8,9]. This initiates ER stress and a unique autophagy [10,11] with subsequent extrusion of an extracellular monocyte-adhesive HA matrix after division that leads to inflammatory responses (Figure 1, Hyperglycemic). We propose that pre-inflammatory monocytes/macrophages (Mpi) recruited into the diabetic tissues initiate inflammatory and fibrotic responses that are ineffective in removing the HA matrix, which mediates the initiation and progression of diabetic complications.
Heparin is a highly sulfated, hence highly polyanionic, glycosaminoglycan with a repeating disaccharide that contains a hexuronic acid (either glucuronic acid or iduronic acid) and glucosamine (either N-acetylated or N-sulfated). It is synthesized as a proteoglycan (serglycin) that is found in mast cells. Daily IP injections of a small amount of heparin in the Streptozotocin (STZ) diabetic rats prevented the pathological responses even though the animals sustained hyperglycemic levels of glucose throughout [12]. This led to clinical trials with heparin for treatment of diabetic patients. However, the molecular and cellular mechanism(s) underlying the roles of heparin are still unclear. Low concentrations (Kd ~20 nM) of heparin prevents the intracellular HA synthesis in hyperglycemic dividing bone marrow progenitor cells and reprograms them to synthesize a monocyte-adhesive extracellular HA matrix after division (Figure 1, Heparin/Hep-Tri) [8,13,14]. We propose that the tissue repairing monocytes/macrophages (Mtr) under hyperglycemia treated with heparin effectively remove the HA matrix without initiating the inflammatory and fibrotic responses, which allows the cells and tissue to maintain their functions while still synthesizing a monocyte-adhesive HA matrix in response to the hyperglycemia. This prevents the cytosolic concentrations of UDP-GlcNAc and UDP-GlcUA from increasing to unacceptable levels during cell division under glucose stress [7].
Hyperglycemia and Hematopoiesis
Hyperglycemia in diabetes induces impairment of hematopoiesis, an important consequence in bone marrow (BM) that contributes to chronic complications in advanced diabetes [15-17]. Preclinical and clinical studies have shown impaired function of hematopoietic stem cells (HSCs) and hematopoietic progenitor cells (HPCs) [15,16,18-20]; of increased numbers of activated inflammatory monocytes/macrophages [15,21-23]; and of accumulation of monocyte adhesive HA matrices in diabetic BM [9]. Euglycemic control in diabetes minimally reverses the HSC and HPC defects [16]. A mechanism of central importance is an increased ratio of Mpi/Mtr monocytes/macrophages in diabetic BM in both animal models and humans [23-25]. This appears to have key roles in inducing inflammatory processes that are poorly understood [15,22,25-27]. Further, anti-inflammatory drugs (NSAIDS) do not reverse diabetic BM dysfunctions. Understanding the formation of the pro-inflammatory Mpi monocytes and macrophages in diabetic BM and the reason for their NSAID resistance is an important goal for future studies [15].
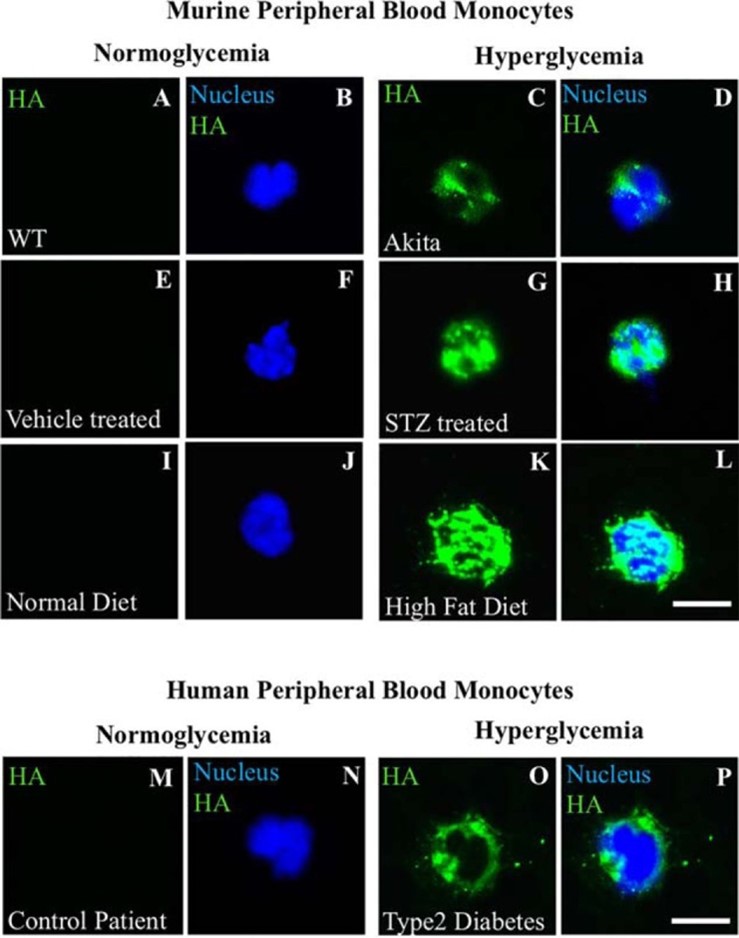
A recent comprehensive review article describes alterations to blood cells associated with DM pathologies [21]. It concludes: “This review expands our current understanding of the role of DM in risk of development of blood cell damages. However, precise mechanism of DM action that stimulates these damages remains unclear.” Further, HA was not featured in any of the cited papers [21]. Our recent publication indicates that aberrant intracellular synthesis of HA by hyperglycemic dividing BM progenitors is the central mechanism involved [13]. It shows that: 1) circulating monocytes of hyperglycemic diabetic patients with hemaglobulin blood A1c >7 have intracellular HA, which is absent in non-hyperglycemic patients with A1c <7; and 2) 3 mouse models for diabetes [Akita, STZ treated, High fat diet] also showed circulating monocytes with intracellular HA only when their blood glucose levels were elevated to diabetic levels (Figure 2). We demonstrated that macrophages that divided in hyperglycemia became pro-inflammatory Mpi and that the presence of low concentrations of heparin (~50 nM) prevented the intracellular HA synthesis and promoted a tissue repair Mtr phenotype (Figures 5 and 6 in [13]). Thus, we hypothesize that hyperglycemia compromises the functions of HSCs and HPCs by initiating the intracellular HA induced ER stress response during their asymmetric cell division, which generates pro-inflammatory Mpi monocytes and prevents generation of anti-inflammatory, tissue-reparative Mtr monocytes.
Future Studies and Conclusion
We are the first research team to determine that intracellular HA is intrinsically involved in the pathological mechanism that occurs in dividing cells stressed by hyperglycemia. Although intracellular HA has been documented in smooth muscle cells cultured in DMEM [28], it was not recognized as a high glucose induced response because DMEM, which is hyperglycemic, has long been used as the standardized medium for cell culture. Through the studies conducted over the past 10 years, we have established the pathological mechanism that occurs in dividing cells stressed by hyperglycemia. Therefore, we propose that the intracellular HA-mediated inflammatory responses in BM HSCs and HPCs are central to many diabetic pathologies. Our discovery of these mechanisms provides the opportunity to develop new approaches for ameliorating some diabetic complications, including impairment and disruption of hematopoiesis [13]. We have shown that heparin interferes with intracellular HA synthesis, ER stress and the induced inflammatory responses in hyperglycemic stressed cells [8,13,29]. Further, the non-reducing terminal trisaccharide of heparin, Hep-Tri, is sufficient to reverse the hyperglycemic stress responses [14]. This is very significant because Hep-Tri lacks the deleterious side effects of heparin. Hep-Tri does not have anti-coagulant activity nor does it bind to a variety of potentially important growth factors [30,31], which have been complicating factors for the use of heparin in the diabetic rat model and in other translational studies that have been tried [12]. Hep-Tri can be used to target hyperglycemic hematopoietic cell alteration by preventing HSC and/or HPC differentiation to pro-inflammatory Mpi monocytes with loss of function, and promoting tissue repair anti-inflammatory Mtr monocytes. Thus, the Hep-Tri cell surface binding receptor and/or its downstream components during division represent a target that may be leveraged to ameliorate the complications of diabetes. We are looking for innovative treatments with Hep-Tri to prevent abnormal high glucose inside dividing cells by activating the hexosamine synthetic pathway (HSP) to synthesize HA that is secreted extracellularly. To best of our knowledge, this will be the first proposed study in the field to utilize the HSP as a tool to detoxify the glucose toxicity from hyperglycemia.
We propose that pro-inflammatory responses induced in monocytes by hyperglycemia are the result of abnormal accumulation of intracellular hyaluronan during their formation by bone marrow progenitor cells. The future studies will be to investigate whether the very large and polyanionic HA accumulates abnormally directly within intracellular compartments, ER, Golgi, transport vesicles, in monocytes and/or leukocytes, BM HSCs and HPCs. Our recent study [13] showed, in three diabetic mouse models and in a human patient, that circulating monocytes contain extensive intracellular HA, and they are expected to be Mpi in most diabetic complications, as shown in our studies [8,9] with kidney glomeruli in the STZ diabetic rat.
Three goals can be pursued in the future studies: 1) Determine the basic mechanisms for how hyperglycemia-induced intracellular metabolic stress in dividing HPCs leads to pro-inflammatory Mpi monocytes, and identify ways to prevent it; 2) Identify which HPC lineages are at greater risk of developing the intracellular HA stress response during division in hyperglycemia; and 3) Test whether heparin and Hep-Tri have therapeutic utility. This objective is based on our discovery [14] that the non-reducing trisaccharide of heparin, Hep-Tri, without anticoagulant activities [30,31], reverses the hyperglycemic stress responses. Diabetic complications associated with hyperglycemia result in extensive and chronic inflammation in numerous organ systems. These proposed studies will reveal new insights regarding the potential therapeutic roles of Hep-Tri to sustain normal hematopoietic cell functions by: 1) preventing formation of Mpi monocytes, and 2) promoting tissue repair Mtr monocyte activation in diabetic bone marrow.
Competing Interests
The authors have no competing interests.
Authors’ Contributions
A.J.W, and V.C.H. prepared and wrote the manuscript.
References
2. Iglay K, Hannachi H, Joseph Howie P, Xu J, Li X, Engel SS, et al. Prevalence and co-prevalence of comorbidities among patients with type 2 diabetes mellitus. Current Medical Research and Opinion. 2016;32(7):1243-52.
3. Davidson JA, Parkin CG. Is Hyperglycemia a Causal Factor in Cardiovascular Disease? Does proving this relationship really matter? Yes. Diabetes Care. 2009;32(suppl 2):S331-S3.
4. Maconi G, Furfaro F, Sciurti R, Bezzio C, Ardizzone S, de Franchis R. Glucose intolerance and diabetes mellitus in ulcerative colitis: pathogenetic and therapeutic implications. World Journal of Gastroenterology. 2014;20(13):3507-15.
5. Hascall VC. The journey of hyaluronan research in the Journal of Biological Chemistry. Journal of Biological Chemistry. 2019;294(5):1690-6.
6. Wang A, de la Motte C, Lauer M, Hascall V. Hyaluronan matrices in pathobiological processes. The FEBS Journal. 2011;278:1412-8.
7. Hascall VC, Wang A, Tammi M, Oikari S, Tammi R, Passi A, et al. The dynamic metabolism of hyaluronan regulates the cytosolic concentration of UDP-GlcNAc. Matrix Biology. 2014;35:14-7.
8. Wang A, Ren J, Wang CP, Hascall VC. Heparin prevents intracellular hyaluronan synthesis and autophagy responses in hyperglycemic dividing mesangial cells and activates synthesis of an extensive extracellular monocyte-adhesive hyaluronan matrix after completing cell division. Journal of Biological Chemistry. 2014;289(13):9418-29.
9. Wang A, Midura RJ, Vasanji A, Wang AJ, Hascall VC. Hyperglycemia diverts dividing osteoblastic precursor cells to an adipogenic pathway and induces synthesis of a hyaluronan matrix that is adhesive for monocytes. Journal of Biological Chemistry. 2014;289(16):11410-20.
10. Ren J, Hascall VC, Wang A. Cyclin D3 mediates synthesis of a hyaluronan matrix that is adhesive for monocytes in mesangial cells stimulated to divide in hyperglycemic medium. Journal of Biological Chemistry. 2009;284(24):16621-32.
11. Wang A, Hascall VC. Hyperglycemia, intracellular hyaluronan synthesis, cyclin D3 and autophagy. Autophagy. 2009;5(6):864-5.
12. Gambaro G, van der Woude FJ. Glycosaminoglycans: use in treatment of diabetic nephropathy. Journal of the American Society of Nephrology. 2000;11(2):359-68.
13. Abbadi A, Loftis J, Wang A, Yu M, Wang Y, Shakya S, et al. Heparin inhibits proinflammatory and promotes anti-inflammatory macrophage polarization under hyperglycemic stress. Journal of Biological Chemistry. 2020;295(15):4849-57.
14. Wang CP, Hascall VC, Zhang F, Linhardt RJ, Abbadi A, Wang A. The responses of hyperglycemic dividing mesangial cells to heparin is mediated by the non-reducing terminal trisaccharide. Journal of Biological Chemistry. 2015;290(48):29045-50.
15. Fadini GP, Ferraro F, Quaini F, Asahara T, Madeddu P. Concise review: diabetes, the bone marrow niche, and impaired vascular regeneration. Stem Cells Translational Medicine. 2014;3(8):949-57.
16. Rodrigues M, Wong VW, Rennert RC, Davis CR, Longaker MT, Gurtner GC. Progenitor cell dysfunctions underlie some diabetic complications. The American Journal of Pathology. 2015;185(10):2607-18.
17. Saki N, Jalalifar MA, Soleimani M, Hajizamani S, Rahim F. Adverse effect of high glucose concentration on stem cell therapy. International Journal of Hematology-Oncology and Stem Cell Research. 2013;7(3):34-40.
18. Mangialardi G, Oikawa A, Reni C, Madeddu P. Bone marrow microenvironment: a newly recognized target for diabetes-induced cellular damage. Endocrine, Metabolic & Immune Disorders – Drug Targets. 2012;12(2):159-67.
19. Adler BJ, Kaushansky K, Rubin CT. Obesity-driven disruption of haematopoiesis and the bone marrow niche. Nature Reviews Endocrinology. 2014;10(12):737-48.
20. Ferraro F, Lymperi S, Mendez-Ferrer S, Saez B, Spencer JA, Yeap BY, et al. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Science Translational Medicine. 2011;3(104):104ra1.
21. Szablewski L, Sulima A. The structural and functional changes of blood cells and molecular components in diabetes mellitus. Biological Chemistry. 2017;398(4):411-23.
22. Fadini GP, Cappellari R, Mazzucato M, Agostini C, Vigili de Kreutzenberg S, Avogaro A. Monocyte-macrophage polarization balance in pre-diabetic individuals. Acta diabetologica. 2013;50(6):977-82.
23. Motyl KJ, Botolin S, Irwin R, Appledorn DM, Kadakia T, Amalfitano A, et al. Bone inflammation and altered gene expression with type I diabetes early onset. Journal of Cellular Physiology. 2009;218(3):575-83.
24. Hu P, Thinschmidt JS, Yan Y, Hazra S, Bhatwadekar A, Caballero S, et al. CNS inflammation and bone marrow neuropathy in type 1 diabetes. The American Journal of Pathology. 2013;183(5):1608-20.
25. McCabe LR. Understanding the pathology and mechanisms of type I diabetic bone loss. Journal of Cellular Biochemistry. 2007;102(6):1343-57.
26. Fornoni A, Ijaz A, Tejada T, Lenz O. Role of inflammation in diabetic nephropathy. Current Diabetes Reviews. 2008;4(1):10-7.
27. Kanter JE, Kramer F, Barnhart S, Averill MM, Vivekanandan-Giri A, Vickery T, et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(12):E715-24.
28. Evanko SP, Wight TN. Intracellular localization of hyaluronan in proliferating cells. Journal of Histochemistry and Cytochemistry. 1999;47(10):1331-42.
29. Wang AJ, Ren J, Abbadi A, Wang A, Hascall VC. Heparin affects cytosolic glucose responses of hyperglycemic dividing mesangial cells. Journal of Biological Chemistry. 2019;294(16):6591-7.
30. Capila I, Linhardt RJ. Heparin-protein interactions. Angewandte Chemie International Edition England. 2002;41(3):391-412.
31. Rabenstein DL. Heparin and heparan sulfate: structure and function. Natural Product Reports. 2002;19(3):312-31.