Abstract
Macrophages, and related cells, are key players of the innate immune response against pathogens. However, they can also be implicated in pathogen persistence and associated pathogenesis. Hereby, we discussed the dual role of liver macrophages during Hepatitis B virus infection. Whereas pro-inflammatory macrophage secretions can inhibit the viral infection, modulation of macrophages phenotype by the virus favour the establishment and maintenance of the infection.
Keywords
Hepatitis B virus, Pro-inflammatory macrophage, Anti-inflammatory macrophage, Immune modulation, Kupffer cells, IL-1beta, Primary cells, Anti-viral
Introduction
Hepatitis B virus (HBV) chronically infects more than 250 million individuals worldwide and is responsible for more than 800,000 deaths per year by promoting end-stage liver diseases, among which decompensated cirrhosis and hepatocellular carcinoma (HCC) (WHO, July 2020) are prominent. Studies performed in chimpanzees or in animalversion of HBV (woodchuck HBV: WHBV) highlighted the lack of immune responses against the virus upon primary infection [1,2]. Thus, HBV has been described as a “stealth” virus (i.e. a virus that does not modify/induce immune response in the cell) [1]. However, a growing number of studies describe that HBV is able to rapidly and efficiently counteract the innate immune response in a large variety of cells (hepatocytes, macrophages, Natural Killer cell…) [3]. Hereby, we focus on the role of macrophages (Mφ) during HBV infection [4].
In the innate immune cell family, Mφ are one of the key components and form one of the first line of defence against pathogens, as they are present at basal level (resident) in many organs. Kupffer cells, the liver resident macrophages, representing 1/5th of total body macrophages, are both constantly exposed to pathogens and play a key role in avoiding systemic infection coming from the enteric circulations [5]. Moreover, upon injury or infection, monocytes can be recruited and differentiate into macrophages to reinforce the local innate immune response. In human, no specific marker has been identified to recognize Kupffer cells from infiltrating monocytes differentiated into macrophage. Thus, macrophages extracted from human liver should be referred as liver macrophages rather than Kupffer cells only.
Mφ can mount different types of response and can be roughly divided into two opposed phenotypes [6]: proinflammatory Mφ (also called M1) and anti-inflammatory Mφ (also called M2). M1-Mφ secretes a variety of inflammatory cytokines such as IL-6 or IL-1β, and are involved in inflammation and pathogens clearance. M2-Mφ secretes IL-10 and TGFβ, among others, and are involved in the resolution of inflammation, anti-parasite responses, and wound healing. M2-Mφ have also been associated with tumour development. Whereas both types have opposed mechanisms of action, they are both mandatory to mount a potent but timely controlled immune response; the return to homeostasis being particularly important in the case of a non-specific (i.e. no specific pathogen epitope involved) response.
Role of M1-Mφ during HBV Infection
M1-Mφ have been largely described to play a role in the immune responses against different viruses [5]. In the case of HBV infection, different pro-inflammatory cytokines have been shown to inhibit both the establishment and maintenance of the infection. Indeed, several studies using recombinant cytokines highlighted the potent effect of pro-inflammatory secretion against HBV infection, with the most potent cytokine being IL-1β [7]. IL-1β was shown to impact on several replication intermediates, with the exception of the circular covalently closed DNA (cccDNA, the viral minichromosome), after one treatment in infectious models [7,8]. Moreover, in our recent study, we observed that pre-treatment with IL-1β strongly inhibits the establishment of the infection in HepaRG, cell culture hepatocytes, and primary human hepatocytes [4]. These results highlight that the local secretion of IL-1β by macrophages in the liver could play a key role in both controlling and preventing the propagation of the infection. Whereas these results served as a proof of concept, more direct evidences of the anti-viral role of M1-Mφ were lacking. Using conditioned media, we were able to show that M1-Mφ secretions impaired the establishment of HBV infection in hepatocytes, highlighting the relevance of previously published data [4]. Of note, the use of a cytokine cocktail produced by Mφ rather than the use of one recombinant cytokine might trigger different pathways, thus leading to complementary anti-viral mechanisms.
Given the anti-viral role of M1-Mφ, HBV infection should theoretically only be productive if the activation of pro-inflammatory response by the resident Kupffer cells, as well as circulating monocytes (that could be recruited to the site of infection) can be avoided. As mentioned before, HBV does not induce strong immune activation, thus preventing the implementation of an inflammatory response upon infection [1]. On the one hand, this is due to a well-evolved life cycle, in which DNA delivery and replication only occurs in the host cell nucleus, thus avoiding a recognition of viral DNA by pattern recognition receptors (PRR). On the other hand, a growing number of studies highlight that HBV is able to inhibit pro-inflammatory innate immune responses. Indeed, we and others showed that upon exposition to cell-supernatant purified HBV, macrophages secreted less IL-1β (as well as IL-6 and TNFα) independently of the type of inflammasome induced to trigger pro-IL-1β processing [4,9,10]. This is a rapid process since an ex vivo overnight exposure to HBV was sufficient to recapitulate an inhibition of IL-1β secretion in TLR4 or AIM2-stimulated liver Mφ. Interestingly, monocyte differentiation into M1- Mφ (mimicking the recruitment of monocytes to the site on infection) was inhibited by the presence of HBV as shown by a strong decrease of IL-1β and IL-6 secretion. In addition, constant exposure to HBV-replicating cells or higher in vitro multiplicity of infection, highlighted that increasing dose of HBV further inhibits M1-Mφ secretions/responses. This indicates that local inhibition, i.e. at proximity of hepatocytes replicating HBV, could be strong, whereas long distance inhibition (systemic) could be lower. Importantly, the HBV-induced inhibition of proinflammatory secretions by M1-Mφ was sufficient to revert the anti-viral effect on the establishment of the infection in hepatocytes [4]. Therefore, HBV might be able to block the local activation of Kupffer cells/liver Mφ, triggered by HBV infection or bacterial components coming from the enteric circulations, to avoid viral decay (Figure 1).
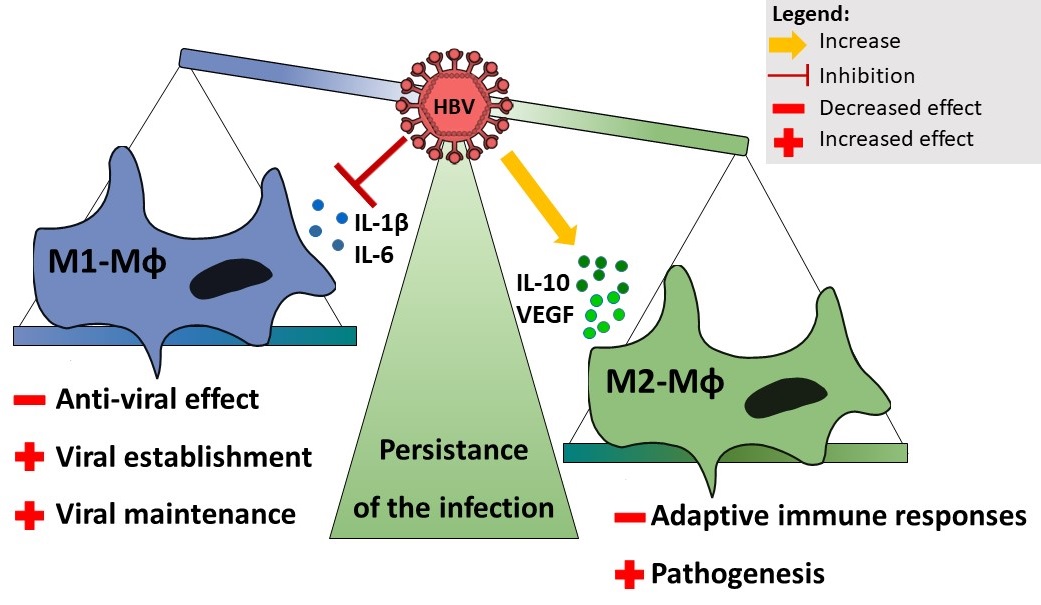
Figure 1. Effect of HBV on macrophages phenotype. HBV inhibits IL-1β and IL-6 secretions by M1-Mφ, preventing anti-viral effect and favouring the establishment and maintenance of the infection. HBV increases anti-inflammatory secretions by M2-Mφ, preventing the implementation of adaptive immune responses and favouring pathogenesis. Mφ: Macrophage; HBV: Hepatitis B virus.
However, other studies have suggested that HBV does not inhibit innate responses. Suslov and colleagues used liver biopsies from HBV infected patients to show that HBV was not able to inhibit the secretion of pro-inflammatory cytokines upon a high dose of different PRR stimulators [11]. Knowing that increasing doses and constant exposure to HBV are leading to increased inhibition, it can be hypothesized that the lack of virus produced in this ex vivo model could account for the discrepancies observed with our model.
Taken together, these data suggest that preventing viral particles and proteins/antigens production, in order to release the viral pressure on Mφ, and then activating innate immune responses, could be the future for the treatment of chronic HBV patients.
Role of M2-Mφ During HBV-infection
The liver can be considered as a tolerogenic organ, given its unique position at the interface between the enteric and systemic circulations. Indeed, the constant exposure to bacterial content coming from the enteric circulations renders the liver macrophages more tolerant, as also observed in our study in which 10 times more TLR4 ligand was mandatory to induce immune response as compared to blood monocytes derived macrophages [4]. Thus, at basal state, it is considered that the liver macrophages are rather in a M2-like Mφ phenotype.
M2-Mφ act as negative regulators of the inflammation, notably by IL-10 and TGFβ secretion [5]. In the case of HBV infection, numerous studies have reported an increase of IL-10 both systemically and locally in the liver during chronic infection [3]. Our results also showed increased IL- 10 production by M2-Mφ when they were exposed to HBV during the stimulation with TLR4 ligands. Moreover, we observed increased CD163+ Mφ in the liver of chronic HBV patients, highlighting more M2-like Mφ in the presence of HBV in vivo [4]. Given that more M2-Mφ are observed in the liver of HBV patients, activation of Mφ locally might trigger IL-10 secretion, reinforcing the immune tolerant environment. Indeed, increased IL-10 secretion by M2-Mφ was sufficient to further inhibit T cell activation [4]. Thus, increased anti-inflammatory secretion locally might play an important role in the inhibition of adaptive immune responses and T-cell exhaustion [12] (Figure 1).
Rendering the liver immune-tolerant and preventing adaptive immune response enables HBV to chronically infect patients for decades without facing potent and specific anti-viral responses. Unfortunately, this phenotype also leads to the development of associated pathogenesis, such as cirrhosis or HCC. M2-Mφ have been associated with disease progression, among which cancer development, in different model [5]. In addition to their anti-inflammatory activity, they secrete other factors important for wound healing, such as vascular endothelial growth factor (VEGF), which are also involved in tumour development. Several studies reported increased VEGF levels during HBV infection [13]. We showed that HBV exposure increased VEGF mRNA in liver- and M2-Mφ [4]. It could be hypothesized that modification of macrophage secretions by HBV might favour HCC development. However, these findings are quite difficult to confirm in vivo as, for the time being, no model has been published recapitulating HBV-associated pathogenesis [14]. In addition, most of the studies in animal models have only looked at the quantity of macrophages rather than the quality of the cells. It is important to keep in mind that HBV infection is not associated with a strong immune response, and thus there should not be a recruitment of monocyte to the liver, whereas the liver Mφ might be influenced towards some specific response by HBV. Of note, our hypothesis refers to chronic HBV infection development as acute infection are associated with strong immune activation.
In chronic HBV patients, this hypothesis on liver Mφ are not easy to confirm due to the scarcity of liver biopsies performed (i.e. the procedure being too invasive compared to the benefice for the patients). However, it will be interesting to assess if the blood monocytes from HBV patients are “pre-disposed” to become M2-like Mφ, due to the exposure to viral particles/proteins systemically. It could be hypothesized that, once the monocytes are taken out of a HBV reached environment, the phenotype is lost, as suggested before. It will be important to understand if inhibiting viral secretions is sufficient to revert the increase of M2-Mφ phenotype in patients, before moving forward with the development of therapeutic strategies targeting the activation of innate immune response.
Altogether, increased M2-Mφ secretions might play an important role in the control of innate and adaptive immune responses as well in the development of associated pathogenesis such as HCC.
Targeting Macrophages to Treat HBV Infection
The use of pro-inflammatory cytokines as a direct treatment for HBV infection remains unrealistic due to severe side effects and cytokine storms that could be expected in vivo. Thus, a different approach should be used to induce a local production of IL-1β. One promising way to trigger pro-inflammatory response locally is the use of PRR agonists. Both in vitro and in vivo, PRR agonists, such as TLR2 or TLR7 or TLR8, have shown potent effect against the infection [15-17]. However, it could be expected that if the delivery/stimulation is not sufficient in the liver, the HBV-induced inhibition might prevent the activation of a strong innate immune response locally. Thus, it will be important (i) to insure efficient delivery of the ligands and strong immune activation locally, and (ii) to test these therapies in combination with direct antiviral agents on the market or in development in order to release the HBV-pressure on innate immune cells before inducing inflammatory responses. This double edge sword (releasing viral pressure and inducing immune response) could be mandatory toward the development of efficient anti-HBV therapies (Figure 2).
Another way to insure a potent M1-Mφ response could be the use of molecules repolarising M2 towards M1 like phenotype (Figure 2). These treatments have two advantages: (i) reducing the number of deleterious M2- Mφ, and (ii) increasing the number of beneficial M1- Mφ. Different treatment have been suggested to be able to repolarise Mφ [18]. Given the role of NF-κB in Mφ polarization, modified liposomes containing NF-κB decoy complexes showed promising results in the conversion of tumour associated M2 into M1-Mφ [18]. Moreover, M1-Mφ have a peculiar metabolism, in which two breaks can be observed in the Krebs cycle, leading to citrate and succinate accumulation [6]. This phenotype renders them independent of the mitochondrial respiration. Thus, inhibition of mitochondrial respiration allows the specific decay of M2-like phenotype and the rise of M1- like phenotype [18]. Additionally, the citrate accumulation observed in the TCA cycle of the M1-Mφ indicates an absence of conversion to α-ketoglutarate. In order to pursue the TCA cycle, M1-Mφ use the α-ketoglutarate coming from glutamine metabolism. Interestingly, increasing glutathione (a derivate of the glutamine metabolism) has shown to favour a M1 response in Mφ [18]. Different molecules have been described to potently increase glutathione in Mφ [18].
Finally, different strategies can be envisaged to reduce M2-Mφ count (Figure 2). Inhibiting the prostaglandin E2, IL-6, and STAT3 pathway activation reduced tumour growth and supressed attraction and differentiation of monocyte into M2-Mφ [19]. Another approach is the use of specific depleting antibody against the colony stimulating factor 1 receptor (CSF1R). Indeed, anti-CSF1R antibody have shown promising results in slowing down cancer progression of different ethology and is currently in phase II studies [20]. Whereas this approach has the advantage to decrease the anti-inflammatory response, it does not per se lead to a potent pro-inflammatory response. Thus, combination with pro-inflammatory stimulators, such as PRR ligand, will be necessary to insure a potent immune response.
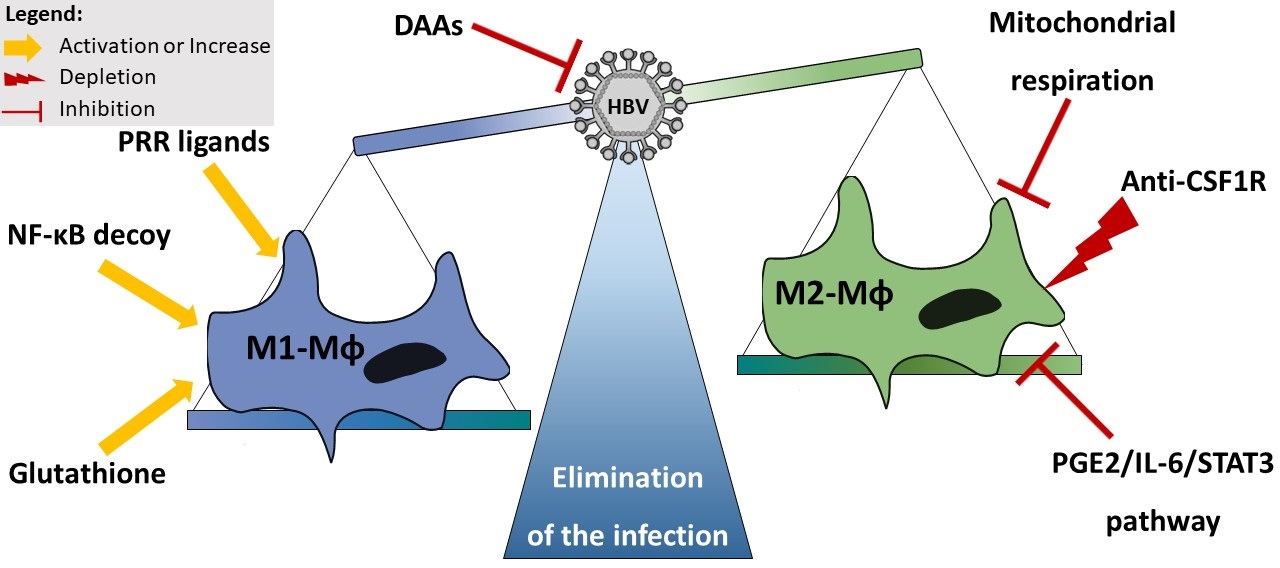
Figure 2. Envisaged treatment to modulate macrophages phenotype. Direct inhibition of HBV by DAAs will release the viral pressure on the Mφ. Activation with PRR ligands or NF-B decoy, and/or Glutathione increase repolarises Mφ towards M1 phenotype. Inhibition of mitochondrial respiration and PG/IL-6/STAT3 pathway, as well as depletion with anti-CSF1R antibody decreases M2-Mφ number. CSF1R: Colony Stimulating Factor 1 Receptor; DAAs: Direct Antiviral Agents; HBV: Hepatitis B Virus Mφ: Macrophage; NF-κB: Nuclear Factor Kappa B; PGE2: Prostaglandin E2; STAT3: Signal Transducer and Activator of Transcription 3.
Conclusion
Mφ play a dual role during HBV infection. On the one hand, M1-Mφ, which are potent anti-viral cells, are inhibited by the virus. On the other hand, M2-Mφ, which are anti-inflammatory/pro-viral cells, are activated by the virus. Thus, targeting macrophages to fight HBV infection could lead to two kills with one stone. In the one hand, inducing pro-inflammatory Mφ secretions should induce strong and potent anti-viral responses. On the other hand, preventing anti-inflammatory Mφ secretions could prevent the inhibition of adaptive immune response and pathogenesis.
Conflicts of Interest
SFD, JL, and DD have no conflicts of interest to declare.
Funding Statement
SFD is supported by the SFBTR179, and JL and DD are supported by grants from the ANRS (French national agency for research on AIDS and viral hepatitis) as well as financial support of INSERM.
Acknowledgments
The authors would like to acknowledge all the persons involved in the original publication [4].
References
2. Michalak TI. Occult persistence and lymphotropism of hepadnaviral infection: insights from the woodchuck viral hepatitis model. Immunological Reviews. 2000 Apr 1;174(1):98-111.
3. Faure-Dupuy S, Lucifora J, Durantel D. Interplay between the hepatitis B virus and innate immunity: from an understanding to the development of therapeutic concepts. Viruses. 2017 May;9(5):95-115.
4. Faure-Dupuy S, Delphin M, Aillot L, Dimier L, Lebossé F, Fresquet J, et al. Hepatitis B virus-induced modulation of liver macrophage function promotes hepatocyte infection. Journal of Hepatology. 2019 Dec 1;71(6):1086-98.
5. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nature Reviews Immunology. 2017 May;17(5):306-21.
6. Hume DA. The many alternative faces of macrophage activation. Frontiers in Immunology. 2015 Jul 22;6:370.
7. Isorce N, Testoni B, Locatelli M, Fresquet J, Rivoire M, Luangsay S, et al. Antiviral activity of various interferons and pro-inflammatory cytokines in non-transformed cultured hepatocytes infected with hepatitis B virus. Antiviral Research. 2016 Jun 1;130:36-45.
8. Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, et al. Interleukin-1 and tumor necrosis factor-α trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). Journal of Biological Chemistry. 2013 Nov 1;288(44):31715-27.
9. Zannetti C, Roblot G, Charrier E, Ainouze M, Tout I, Briat F, et al. Characterization of the inflammasome in human kupffer cells in response to synthetic agonists and pathogens. The Journal of Immunology. 2016 Jul 1;197(1):356-67.
10. Yu X, Lan P, Hou X, Han Q, Lu N, Li T, et al. HBV inhibits LPS-induced NLRP3 inflammasome activation and IL-1β production via suppressing the NF-κB pathway and ROS production. Journal of Hepatology. 2017 Apr 1;66(4):693-702.
11. Suslov A, Boldanova T, Wang X, Wieland S, Heim MH. Hepatitis B virus does not interfere with innate immune responses in the human liver. Gastroenterology. 2018 May 1;154(6):1778-90.
12. Bertoletti A, Ferrari C. Adaptive immunity in HBV infection. Journal of Hepatology. 2016 Apr 1;64(1):S71-83.
13. Alkharsah KR. VEGF upregulation in viral infections and its possible therapeutic implications. International Journal of Molecular Sciences. 2018 Jun;19(6):1642.
14. Dandri M, Petersen J. Animal models of HBV infection. Best Practice & Research Clinical Gastroenterology. 2017 Jun 1;31(3):273-9.
15. Niu C, Li L, Daffis S, Lucifora J, Bonnin M, Maadadi S, et al. Toll-like receptor 7 agonist GS-9620 induces prolonged inhibition of HBV via a type I interferondependent mechanism. Journal of Hepatology. 2018 May 1;68(5):922-31.
16. Lucifora J, Bonnin M, Aillot L, Fusil F, Maadadi S, Dimier L, et al. Direct antiviral properties of TLR ligands against HBV replication in immune-competent hepatocytes. Scientific Reports. 2018 Mar 29;8(1):5390.
17. Mackman RL, Mish M, Chin G, Perry J, Appleby TC, Aktoudianakis V, et al. Discovery of GS-9688 (Selgantolimod), as a Potent and Selective Oral Tolllike Receptor 8 Agonist for the Treatment of Chronic Hepatitis B. Journal of Medicinal Chemistry. 2020 May 14;63:10188–203.
18. Fraternale A, Brundu S, Magnani M. Polarization and repolarization of macrophages. J Clin Cell Immunol. 2015;6(319):2.
19. Heusinkveld M, van der Burg SH. Identification and manipulation of tumor associated macrophages in human cancers. Journal of Translational Medicine. 2011 Dec 1;9(1):216.
20. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. Journal for Immunotherapy of Cancer. 2017 Dec 1;5(1):53.