Abstract
This study reports sequence data mining and analysis, complete coordinate tertiary structure prediction including Deep Learning inspired validation, and in silico functional characterization of the full SARS-CoV-2 proteome based on the NCBI reference sequence NC_045512 (29903 bp ss-RNA). Out of 25 polypeptides analyzed, 3D structures of 15 of them were predicted using comparative protein structure prediction method and ab-initio modelling method due to unavailability of experimentally determined structures. Deep Learning and Neural Network based tools such as QMEANDisCo 4.0.0, MolProbity 4.4, ProQ3D and Procheck were used to verify the predicted 3D structures. Tunnel analysis revealed the presence of multiple tunnels in NSP4, nucleocapsid phosphoprotein, NSP3, membrane glycoprotein, ORF6 protein, NSP1, NSP6, and envelop protein, indicating a large number of transport pathways for small ligands that influence their reactivity. Ligand-binding pockets with high estimates of druggability scores were detected in envelope glycoprotein (0.97), membrane glycoprotein (0.87), NSP6 (0.79), ORF7a (0.79), ORF8 (0.75), ORF3a (0.72), and NSP4 (0.70), indicating the ability to bind drug-like molecules with high affinity indicating that the predicted structures would be useful for protein nanotechnology in understanding protein machinery towards drug repurposing and discovery studies. Moreover, the molecular phylogenetic analysis of orf1ab polyprotein indicates close relatedness of SARS-CoV-2 to the bat coronavirus.
Keywords
Proteome analysis, Protein machinery, Protein nanotechnology, SARS-CoV-2, Tunnel analysis, Deep learning, Neural network
Highlights
• In silico sequence-based and structure-based functional characterization of the full SARS-CoV-2 proteome
• Reports sequence data mining and analysis, complete coordinate tertiary structure prediction and Machine Learning inspired validation of SARS-CoV-2 proteins
• Ligand-binding pockets with high estimates of druggability scores are analyzed from SARS-CoV-2 proteins
• Identified the SARS-CoV-2 proteins with high reactivity through tunnel analysis
• Evolutionary analysis of SARS-CoV-2 orf1ab polyprotein indicates close relatedness to the bat coronavirus
Abbreviations
AlphaCoVs: Alpha Coronaviruses; BetaCoVs: Beta Coronaviruses; CoV: Coronavirus; COVID-19: Coronavirus disease 2019; GMQE: Global Model Quality Estimates; MERS-CoV: Middle East Respiratory Syndrome Coronavirus; NCBI: National Center for Biotechnology Information; SARS-CoV-2: Severe Acute Respiratory Syndrome Coronavirus 2
Introduction
Severe acute respiratory syndrome coronavirus 2 (SARSCoV- 2) is a positive-sense, single-stranded RNA with genome size 26.2, and 31.7 kb coronavirus, covered by an enveloped structure [1], which is a major source of disaster in the 21st century. A typical CoV contains at least six ORFs in its genome. SARS-CoV-2 is the seventh coronavirus that is known to cause human disease. Previously identified human CoVs that cause human disease include the alphaCoVs (hCoV-NL63 and hCoV-229E) and the betaCoVs (HCoV-OC43, HKU1, severe acute respiratory syndrome CoV, and Middle East respiratory syndrome CoV). Among these seven strains, three strains proved to be highly pathogenic (SARS-CoV, MERS-CoV, and 2019- nCoV), which caused endemic of severe CoV disease [2-5].
SARS-CoV-2 is an enveloped virus with ≈100 nm in diameter. The role of nanotechnology is highly relevant to counter this “virus” nano enemy. Understanding the complete proteome of SARS-CoV-2 is the need of the hour for the nano intervention in designing effective nanocarriers to counter the conventional limitations of antiviral and biological therapeutics. The major structural proteins, namely the E and M proteins, which form the viral envelope; the N protein, which binds to the virus’s RNA genome, and the S protein, which binds to human receptors, may be significant from the perspective of drug design and development. The nonstructural proteins are expressed as two long polypeptides, which are chopped up by the virus’s main protease. This group of proteins includes the main protease (Nsp5), and RNA polymerase (Nsp12) has equal importance for structure-based drug design. In this regard, the present study reports sequence analysis and structure prediction (both comparative and ab initio modeling) of the full SARS-CoV-2 proteome based on the NCBI reference sequence NC_045512 (29903 bp ss-RNA), which is identical to the GenBank entries MN908947 and MT415321 (Supplementary Table 1). Further, the study included evolutionary analysis of the largest protein of SARS-CoV-2 i.e., orf1ab polyprotein, to understand the evolutionary profile of the SARS-CoV-2.
The SARS-CoV-2 genome encodes 29 proteins. The present analysis included the 25 encoded proteins of NCBI reference genome sequence NC_045512 (Figure 1) (including 15 proteins of orf1ab), of which 15 proteins have not yet been experimentally characterized, and 10 have experimental structures with known PDB IDs. As the study was based on the proteome encoded by the NCBI reference genome (Supplementary Table 1), the proteins, which are either unexpressed or accessory proteins like protein 9b and ORF 14 and not reported in the reference genome, are not included in the present analysis [6].
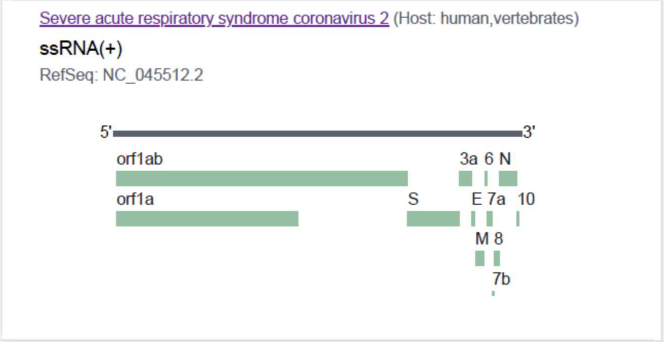
Figure 1. Genome organization of SARS CoV-2 reference genome (NC_045512.2) used in the present study.
Structure-based drug design focuses on the search, design, and optimization of a small molecule that fits well into the binding pocket of a target protein to form energetically favorable interactions. Determination of protein structure by means of experimental methods such as X-ray crystallography or NMR spectroscopy is time consuming and not successful with all proteins, especially with membrane proteins [7]. Knowledge of the 3D structures of proteins provides invaluable insights into protein machinery for the molecular basis of their functions. With the availability of structural information of protein targets, nanotechnologies could bring dramatic increases in the sensitivity of detection technology for research and diagnostic applications, greater selectivity for drug delivery, and detailed insight into biological mechanisms and systems. Lack of knowledge of the 3D structures of most of the proteins of SARS-CoV-2 has hindered such efforts to understand the binding specificities of ligands with protein. A homology modeling method is applied when there is a sufficient amount of similarity between the protein (structure to be predicted) and the template (whose structure has already been determined). However, in the other case, when the similarity between the two is quite low, then the ab-initio method is applied.
Materials and Methods
Acquisition and analysis of the sequences
The sequence of the full SARS-CoV-2 proteome based on the NCBI reference NC_045512 (29903 bp ss-RNA) along with the GenBank entries MN908947 and MT415321 were analyzed (Supplementary Table 1). A total of 25 proteins from the proteome were analyzed, of which 15 proteins had no experimental structures, based on BLASTp [8] and FASTA [9] searches. The amino acid sequence of orf1ab polyprotein (27 nos.) from different coronaviruses with UniProtKB/TrEMBL-IDs YP009724389, A0A4Y5QL57, A0A6G6A323, A0A6G6A2Q6, A0A6B9WIQ1, Q3ZTF4, P0C6Y0, P0C6X4, P0C6Y5, P0C6W9, P0DTD1, P0C6W7, P0C6X7, P0C6X8, P0C6W6, P0C6V9, P0C6X9, K9N7C7, Q98VG9, P0C6X5, P0C6W2, P0C6W8, A0A1S6KXQ5, X2GG12, B1PHI6, C9DSU6, and A0A0A7UXR0 were retrieved for construction protein sequence-based phylogeny.
Comparative and ab-initio modeling
3D structure predictions were carried out for fifteen (15) proteins, which are without 3D coordinate files. BlastP and FASTA searches were performed independently to know the existing structure from the PDB, for a suitable template for comparative modeling, and to select the proteins for which ab-initio modeling is required (Supplementary Table 2). The significance of the BLAST results was assessed and on the basis of e-value generated by the BLAST family of search algorithm, percentage of sequence identity, and query coverage. For comparative modeling sequence identity of >25% and good query coverage was considered for the selection of templates. The ab-initio method was preferred for structure prediction when there was no or very low amount of similarity for the protein. Six (6) proteins, namely NSP1, surface glycoprotein, envelope protein, membrane glycoprotein, ORF7a, and nucleoproteins were predicted following the Modeller9.24 program [10], SWISS-MODEL [11], and Baker Rosetta Server (https://robetta.bakerlab.org); nine (09) proteins, namely NSP2, NSP3, NSP4, NSP6, ORF3a, ORF6, ORF7b, ORF8 and ORF10 having no similarity to the available PDB structures, were modeled using abinitio modelling protocol of Baker Rosetta Server. The loop regions were modeled using the ModLoop server [12]. The final 3D structures with complete coordinates were obtained by optimization of the molecular probability density function of Modeller 9.24 with the variable target function procedure [13].
The computational protein structures were verified by using global model quality estimates (GMQE) and local quality estimates using Neural Network-based QMEANDisCo 4.0.0 and Deep Learning- based ProQ3D [14], MolProbity version 4.4 [15], ProQ (LGscore and MaxSub scores), PROCHECK [16]. A good quality model generally means a QMEAN Z-scores around zero (scores below -4 are an indication of low-quality structure), a lower MolProbity score, a LGscore of >3, a MaxSub score >0.5, and over 90% residues in the most favored regions in the Ramachandran plot. The global model quality estimate (GMQE), which is scaled between 0 and 1 have also been considered during model evaluation. All the graphic presentations of the 3D structures were prepared using the Chimera version 1.8.1 [17]. The final protein coordinate files were analyzed using PROMOTIF program that provides details of the location and types of structural motifs in protein structure by analysis of PDB files [18].
Proteomics analysis
Proteomics analyses were carried out using ExPASy proteomic tools (https://www.expasy.org/tools). The data mining and sequence analyses of the physicochemical parameters of SARS-CoV-2 proteomes were computed using ProtParam [19] and BioEdit [20].
Sequence-based functional annotation was carried out for the SARS-CoV-2 proteome in the sequence database, using Pfam (pfam.sanger.ac.uk/-), and GO (www.geneontology. org/). The ProFunc server [21] was used to identify the likely biochemical function of proteins from the predicted 3D structure. PFam, PROSITE, PRINTS, and InterProScan were deployed for functional characterization. The MOLE 2.0 [22], and the Caver Web 1.0 [23] were used for advanced analysis of bio-macromolecular channels. The theoretical structures of the present study were used for tunnel analysis using Caver Web 1.0. The pocket with the highest relevance score, largest volume (Å3), and highest estimated druggability was considered for tunnel calculation. The tunnel bottleneck radius and length were calculated in Ångström (Å), and throughput (estimated tunnel importance) calculated as e-cost, where e is Euler’s number. Throughput values ranges from 0 to 1; the higher the value, the greater the importance of the pathway [23].
Evolutionary analysis of orf1ab polyprotein
The amino sequences of orf1ab polyprotein of coronaviruses (27 nos.) from different hosts were aligned using ClustalW 1.6 [24] integrated in MEGA X software [25]. The evolutionary history was inferred using maximum likelihood methods, and Le Gascuel model [26]. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates). Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the JTT model, and then selecting the topology with superior log likelihood value. There was a total of 7593 positions in the final dataset.
Results
Tertiary structures of SARS-CoV-2 proteins
Structures predicted using comparative modeling
Leader protein (NSP1): The nonstructural protein NSP1 (IPR021590) is the N-terminal cleavage product of the viral replicase that mediates RNA replication and processing. ProMotif results revealed that the structure of NSP1 protein has 2 sheets, 1 beta hairpin,3 beta bulges,6 strands, 8 helices,4 helix-helix interactions, 19 beta turns, and 8 gamma turns (Table 1; Figures 2A and Supplementary Figure 1). Physicochemical parameter analysis computed that NSP1 has a theoretical isoelectric point (pI) of 5.36, an instability index of 28.83, an aliphatic index of 89.72, and a grand average of hydropathicity of - 0.378 (Supplementary Table 3).
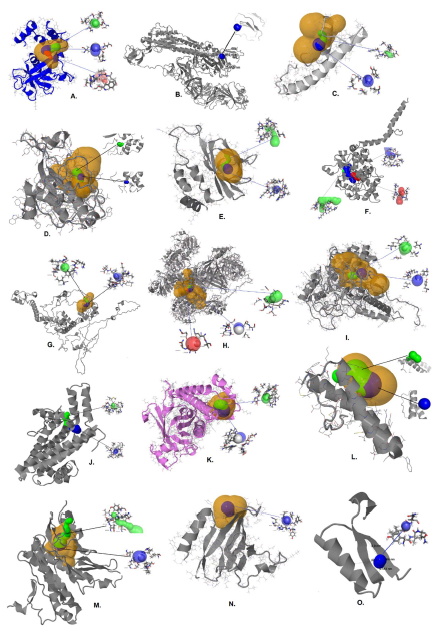
Figure 2. The 15 predicted protein structures of SARS-CoV-2 proteome along with estimated tunnels. A. NSP1, B. Surface Glycoprotein, C. Envelope protein, D. Membrane glycoprotein, E. ORF7a protein, F. Nucleocapsid phosphoprotein, G. NSP2, H. NSP3, I. NSP4, J. NSP6, K. ORF3a protein, L. ORF6 protein, M. ORF 7b protein, N. ORF 8 protein, O. ORF10 protein. Tunnels are colored on the basis of preferences of throughout values i.e., tunnel-1 (blue), tunnel-2 (green), and tunnel-3 (red). The high relevance pockets are shown (yellow).
| Sl. No. |
Protein structure | GMQE | QMEAN4 Z-score |
Mol- Probity Score | Ramachandran Favored regions [%] | ProQ LG- score |
ProQ Max- Sub | Pro- Q3D-TM | Model Archive IDs |
|---|---|---|---|---|---|---|---|---|---|
| 1 | NSP1 | 0.72 ± 0.06 | -1.14 | 1.13 | 94.94% | 4.35 | 0.21 | 0.51 | ma-rt- n7y |
| 2 | surface glycoprotein | 0.71 ± 0.05 | -0.59 | 1.02 | 95.20% | 4.75 | 0.36 | 0.30 | ma- jb7z3 |
| 3 | Envelope protein | 0.50 ± 0.11 | -2.94 | 1.16 | 92.90% | 1.41 | 0.16 | 0.41 | ma- g632o |
| 4 | Membrane glycoprotein | 0.49 ± 0.06 | -1.14 | 1.78 | 89.64% | 5.66 | 0.30 | 0.21 | ma- sgmep |
| 5 | ORF7a protein | 0.63 ± 0.08 | -2.09 | 0.94 | 94.5% | 2.33 | 0.17 | 0.55 | ma- tln95 |
| 6 | Nucleocapsid phosphoprotein | 0.50 ± 0.05 | -0.87 | 0.93 | 94.72% | 1.91 | 0.15 | 0.21 | ma- l97cg |
| 7 | NSP2 | 0.45 ± 0.05 | -1.46 | 1.54 | 93.6% | 3.91 | 0.41 | 0.26 | ma-u9iji |
| 8 | NSP3 | 0.55 ± 0.05 | -1.66 | 0.98 | 91.4% | 3.95 | 0.33 | 0.30 | ma-wy- cyn |
| 9 | NSP4 | 0.49 ± 0.05 | -2.24 | 1.83 | 90.9% | 5.66 | 0.25 | 0.50 | ma- 4msew |
| 10 | NSP6 | 0.58 ± 0.05 | -2.52 | 0.76 | 99.31% | 5.98 | 0.36 | 0.41 | ma- qh7ay |
| 11 | ORF3a protein | 0.71 ± 0.05 | -2.08 | 0.80 | 91.9% | 2.56 | 0.23 | 0.32 | ma-tf3tt |
| 12 | ORF6 protein | 0.65 ± 0.11 | -0.41 | 0.96 | 98.31% | 2.31 | 0.26 | 0.51 | ma- w31qn |
| 13 | ORF7b protein | 0.62 ± 0.12 | -1.04 | 0.59 | 95.7% | 1.5 | 0.11 | 0.50 | ma- wjf4x |
| 14 | ORF8 protein | 0.71 ± 0.08 | -0.64 | 1.03 | 90.76.7% | 2.02 | 0.13 | 0.67 | ma- hnbpo |
| 15 | ORF10 protein | 0.39 ± 0.12 | -0.96 | 1.29 | 94.44% | 1.71 | 0.19 | 0.41 | ma-os- j6n |
| Referenced ranges of quality in ProQ: Correct: LGscore >1.5, MaxSub >0.1; Good: LGscore >3, MaxSub >0.5; Very good: LGscore >5, MaxSub >0.8; GMQE -Global Model Quality Estimation. | |||||||||
Of the eight possible pockets of NSP1, the pocket with relevance score (90%), volume 583 Å3 and estimated druggability 0.54 was considered for tunnel analysis. The high-relevance pocket showed 3 tunnels at a throughput value range of 0.63--0.91. Of the three most potential tunnels, the best tunnel found by CAVER 3.01 geometrical algorithms i.e., tunnel 1(blue) was with the bottleneck radius of 1.6 Å, length of 2.8 Å, distance to surface of 2.6 Å, curvature of 1.1, throughput of 0.91, and number of residues 11; tunnel 2 (green) with bottleneck radius of 2.2 Å, length of 5.3 Å, distance to surface of 5.0 Å, curvature of 1.1, throughput of 0.89, and number of residues 18. Tunnel 3 (red) with bottleneck radius of 0.9 Å, length of 7.6 Å, distance to surface of 5.6 Å, curvature of 1.4, throughput of 0.64, and number of residues 13 (Figure 2A).
Surface glycoprotein (spike glycoprotein): The surface glycoprotein (spike glycoprotein) contains 13 sheets, 18 beta hairpins, 18 beta bulges, 52 strands, 22 helices, 29 helix-helix interactions, 76 beta turns, 16 gamma turns, and 12 disulfides (Table 1; Figures 2B and Supplementary Figure 1). The surface glycoprotein (length=1273 amino acids; molecular weight=141.113kDa) is rich in leucine (8.48%) and serine (7.78) %). The surface glycoprotein had a pI of 6.32, an instability index of 32.86, an Aliphatic index of 84.67, and a grand average of hydropathicity of -0.077 (Supplementary Figure 3A; Supplementary Table 3).
Of the top ten possible pockets of surface glycoprotein, the pocket with relevance score (100%), volume 4784 Å3 and estimated druggability 0.10 was considered for tunnel analysis. The high-relevance pocket showed a single tunnel at a throughput value of 0.81. The estimated high throughput tunnel 1 (blue) in the surface glycoprotein is with a bottleneck radius of 1.1 Å, length of 4.9 Å, distance to surface of 3.0 Å, curvature of 1.7, throughput of 0.81, and number of residues of 7 (Figure 2B).
Envelope protein (E protein): Envelope protein (E protein) has 3 helices, 1 helix-helix interaction, 3 beta turns, and 1 gamma turn (Table 1; Figures 2C and S1). Envelope protein (mw = 8364.59 Da) is rich in leucine (18.67%) and valine (17.33%). The envelope protein had a pI of 8.57, an instability index of 38.68, an Aliphatic index of 144.00, and a grand average of hydropathicity of 1.128 (Supplementary Figure 3B; Supplementary Table 3).
Of the two possible pockets of envelope protein, the pocket with a relevance score (100%), volume of 906Å3 and estimated druggability of 0.97 was considered for tunnel analysis. The high-relevance pocket showed three tunnels at a throughput value range of 0.54-0.92. The estimated most potential high throughput tunnel-1 (blue) is with a bottleneck radius of 1.8 Å, length of 2.5 Å, distance to surface of 2.3 Å, curvature of 1.1, throughput of 0.92, and number of residues 10. Tunnel-2 (green) is with a bottleneck radius of 1.3 Å, length of 5.9 Å, distance to surface of 4.7 Å, curvature of 1.2, throughput of 0.73, and number of residues 13 (Figure 2C).
Membrane glycoprotein (M protein): Membrane glycoprotein has 9 helices, 8 helix-helix interactions, 28 beta turns, and 11 gamma turns (Table 1; Figures 2D and Supplementary Figure 1). Membrane glycoprotein (mw=25145.16 Da) is rich in leucine (15.77%) and isoleucine (9.01%). Membrane glycoprotein presented a pI of 9.51, an instability index of 39.14, an Aliphatic index of 120.86, and a grand average of hydropathicity of 0.446 (Supplementary Figure 3C; Supplementary Table 3).
Of the top ten possible pockets of membrane glycoprotein, the pocket with relevance score (100%), volume 1543 Å3 and estimated druggability of 0.87 was considered for tunnel analysis. The high-relevance pocket showed 5 tunnels at a throughput value range of 0.51-0.89.The two high throughput tunnels are tunnel 1 (blue), with a bottleneck radius of 1.7 Å, length of 3.9 Å, distance to surface of 3.3 Å, curvature of 1.2, throughput of 0.87, and number of residues 17; tunnel-2 (green) is with a bottleneck radius of 1.5 Å, length of 7.4 Å, distance to the surface of 6.4 Å, curvature of 1.2, throughput of 0.76, and number of residues 18 (Figure 2D).
ORF7a protein: Protein 7a (X4 like protein) is a nonstructural protein that is dispensable for virus replication in cell culture. Structurally, it consists of 2 sheets, 2 beta hairpins, 3 beta bulges, 7 strands, 4 helices, 4 helix-helix interactions, 13 beta turns, 5 gamma turns, and 2 disulphides (Table 1; Figures 2E and Supplementary Figure 1). ORF7a protein (mw=13743.47 Da) is rich in leucine (12.40%), threonine (8.26 %), and phenylalanine (8.26%). ORF7a protein has shown a pI of 8.23, an instability index of 48.66, an aliphatic index of 100.74, and a grand average of hydropathicity of 0.318 (Figures S3D; Supplementary Table 3).
Of the four possible pockets of ORF 7a protein, the pocket with relevance score (99%), volume 775 Å3 and estimated druggability 0.79 was considered for tunnel analysis. The high-relevance pocket showed two tunnels at a throughput value range of 0.40-0.91. Tunnel 1 (blue) has a bottleneck radius of 2.1 Å, length of 2.0 Å, distance to surface of 2.0 Å, curvature of 1.0, throughput of 0.91, and number of residues 15. Tunnel 2 (green) with a bottleneck radius of 0.9 Å, length of 15.4 Å, distance to the surface of 12.0 Å, curvature of 1.3, throughput of 0.40, and number of residues 24 (Figure 2E).
Nucleocapsid phosphoprotein: Nucleocapsid phosphoprotein has demonstrated 1 sheet, 1 beta hairpin, 2 strands, 30 helices, 27 helix-helix interactions, 31 beta turns, and 11 gamma turns (Table 1; Figures 2F and Supplementary Figure 1). The nucleocapsid phosphoprotein (mw=45623.27 Da) is rich in glycine (10.26%), alanine (8.83%), and serine (8.83%), along with a pI of 10.07, an instability index of 55.09, an aliphatic index of 52.53, and a grand average of hydropathicity of -0.971 (Supplementary Figure 3E; Supplementary Table 3). Of the top ten possible pockets of the nucleocapsid phosphoprotein, two pockets with a relevance score of 100% and 45% were observed; with pocket-1 containing volume 4861 Å3 and estimated druggability 0.10, and pocket-2 containing volume 3082 Å3 and estimated druggability of 0.46 were detected. The high-relevance pocket showed 7 tunnels in the throughput range of 0.44- -0.70. The high-relevance pocket showed The best tunnel found by CAVER 3.01 geometrical algorithms i.e. tunnel 1(blue) was a bottleneck radius of 2.0 Å, length of 19.6 Å, distance to surface of 14.9 Å, curvature of 1.3, throughput of 0.70, and number of residues 40 ; tunnel 2 (green) with a bottleneck radius of 1.8 Å, length of 25.1 Å, distance to surface of 15.7 Å, curvature of 1.6, throughput of 0.63, and number of residues 45; tunnel 3 (red) with a bottleneck radius of 1.3 Å, length of 20.0 Å, distance to surface of 8.1 Å, curvature of 2.5, throughput of 0.58, and number of residues 34 (Figure 2F).
Structures predicted using ab-initio modeling
Nonstructural protein 2 (NSP2): ProMotif evaluation demonstrated that NSP2 had 2 sheets, 1 beta hairpin, 1 beta bulge, 4 strands, 25 helices, 18 helixhelix interactions, 77 beta turns, 26 gamma turns, and 1 disulfide (Table 1; Figures 2G and S1). NSP2 had a pI of 6.25, an instability index of 36.06, an Aliphatic index of 88.93, and a grand average of hydropathicity of − 0.062 (Supplementary Table 3). This protein may play a role in the modulation of the host cell survival signaling pathway by interacting with host PHB and PHB2.
Of the top ten possible pockets of NSP2, the pocket with relevance score (100%), volume 892 Å3 and estimated druggability of 0.31 was considered for tunnel analysis. The high-relevance pocket showed 2 tunnels at a throughput value range of 0.65--0.82. Tunnel 1(blue) with a bottleneck radius of 1.5 Å, length of 5.4 Å, distance to surface of 4.8 Å, curvature of 1.1, throughput of 0.82, and number of residues 19. Tunnel 2 (green) with a bottleneck radius of 1.1 Å, length of 7.2 Å, distance to surface of 6.6 Å, curvature of 1.1, throughput of 0.65, and number of residues 20 (Figure 2G).
Nonstructural protein 3 (NSP3): The structure of NSP3 has 15 sheets, 5 beta-alpha-beta units, 13 beta hairpins, 2 psi loops, 9 beta bulges, 47 strands, 72 helices, 71 helix-helix interactions, 249 beta turns, 80 gamma turns, and 3 disulfides (Table 1; Figures 2H and Supplementary Figure 1). NSP3 with pI 5.56 has an instability index of 36.56, an Aliphatic index of 86.22, and a grand average of hydropathicity of − 0.175 (Supplementary Table 3).
Of the top ten possible pockets of NSP3, the pocket with relevance score (100%), volume 7919Å3 and estimated druggability 0.51 was considered for tunnel analysis. The high-relevance pocket showed 5 tunnels at a throughput value range of 0.73--0.94. Tunnel 1(blue) with bottleneck radius of 2.8 Å, length of 1.5 Å, distance to surface of 1.4 Å, curvature of 1.1, throughput of 0.94, and number of residues 13. Tunnel 2 (green) with bottleneck radius of 2.1 Å, length of 6.8 Å, distance to surface of 6.4 Å, curvature of 1.1, throughput of 0.87, and number of residues 18. Tunnel 3 (red) with a bottleneck radius of 1.6 Å, length of 5.3 Å, distance to surface of 5.0 Å, curvature of 1.1, throughput of 0.85, and number of residues 14 (Figure 2H).
Nonstructural protein 4 (NSP4): The structure of NSP4 has 2 sheets, 2 beta hairpins, 4 strands, 15 helices, 22 helix-helix interactions, 30 beta turns, 18 gamma turns, and 3 disulphides (Table 1; Figures 2I and S1). NSP4 has a pI of 7.16, an instability index of 34.09, an Aliphatic index 95.50, and a grand average of hydropathicity of 0.343 (Supplementary Table 3).
Of the top ten possible pockets of NSP4, the pocket with relevance score (100%), volume 3961 Å3 and estimated druggability of 0.70 was considered for tunnel analysis. The high-relevance pocket showed 7 tunnels at a throughput value range of 0.23-0.94. Tunnel 1(blue) with a bottleneck radius of 2.0 Å, length of 2.3 Å, distance to surface of 2.2 Å, curvature of 1.0, throughput of 0.94, and number of residues 15. Tunnel 2 (green) with bottleneck radius of 1.8 Å, length of 9.2 Å, distance to surface of 6.1 Å, curvature of 1.5, throughput of 0.82, and number of residues 19 (Figure 2I).
Nonstructural protein 6 (NSP6): NSP6 presented 1 sheet, 1 beta hairpin, 2 strands, 14 helices, 31 helix-helix interactions, 9 beta turns, and 2 gamma turns (Table 1; Figures 2J and S1). NSP6 has a pI value of 9.11, an instability index of 22.94, an Aliphatic index of 111.55 and a grand average of hydropathicity of 0.790 (Supplementary Table 3). Nsp6 may play a role in the initial induction of autophagosomes from the host’s endoplasmic reticulum.
Of the top ten possible pockets of NSP6, the pocket with a relevance score of 100%, volume of 562 Å3 and estimated druggability of 0.64 was considered for tunnel analysis. However, another pocket with a lower relevance score of 53% and volume of 808 Å3 showed a high estimate of druggability of 0.79. The high-relevance pocket showed 3 tunnels at a throughput value range of 0.30--0.81. Tunnel 1 (blue) with bottleneck radius of 1.8 Å, length of 7.8 Å, distance to surface of 7.1 Å, curvature of 1.1, throughput of 0.81, and number of residues 20. Tunnel 2 (green) with bottleneck radius of 1.4 Å, length of 8.7 Å, distance to surface of 7.0 Å, curvature of 1.3, throughput of 0.75, and number of residues 20 (Figure 2J).
ORF3a protein: ORF3a protein (papain-like protease) has 2 sheets, 4 beta hairpins, 1 beta bulge, 8 strands, 12 helices, 12 helix-helix interactions, 13 beta turns, 2 gamma turns, and 1 disulfide (Table 1; Figures 2K and S1). ORF3a protein (mw=31121.29 Da) is rich in leucine (10.91%), valine (9.09%), threonine (8.73%), and serine (8.00%). ORF3a protein has a theoretical pI of 5.55, an instability index of 32.96, an Aliphatic index of 103.42, and a grand average of hydropathicity of 0.275 (Supplementary Figure 3F; Supplementary Table 3).
The potential pocket of ORF3a with a relevance score of 100%, volume of 590Å3 and estimate druggability of 0.72 was considered for tunnel analysis. The pocket showed two tunnels with a throughput value range of 0.92-0.95. The best tunnel found by CAVER 3.01 geometrical algorithms i.e., tunnel 1(blue) with a bottleneck radius of 3.1 Å, length of 3.0 Å, distance to surface of 3.0Å, curvature of 1.0, throughput of 0.95, and number of residues 15; tunnel 2 (green) with a bottleneck radius of 2.7 Å, length of 3.0 Å, distance to surface of 2.6 Å, curvature of 1.1, throughput of 0.92, and number of residues 14 (Figure 2K).
ORF6 protein: ORF6 protein shows 3 helices, 1 helixhelix interaction, 2 beta turns, and 1 gamma turn (Table 1; Figures 2L and S1). ORF6 protein (mw=7272.15 Da) is rich in isoleucine (16.39%) and leucine (13.11%). ORF6 with a pI of 4.60, an instability index of 31.16, an aliphatic index of 130.98, and a grand average of hydropathicity of 0.233 (Supplementary Figure 3G; Supplementary Table 3).
A single pocket with a relevance score of 100%, volume of 840 Å3 and poor estimated druggability of 0.03 was observed in ORF6 protein. The pocket showed 4 tunnels with a throughput value range of 0.69--0.97. Two of the high-throughput tunnels are tunnel 1 (blue) with a bottleneck radius of 1.6 Å, length of 12.7 Å, distance to surface of 8.7 Å, curvature of 1.0, throughput of 0.78, and number of residues 17. Tunnel 2 (green) was with a bottleneck radius of 1.2 Å, length of 7.6 Å, distance to surface of 6.6 Å, curvature of 1.1, throughput of 0.75, and number of residues 12 (Figure 2L).
ORF 7b protein: The structure of ORF7b has 2 helices, 1 helix-helix interaction, 1 beta turns, and 1 gamma turn (Table 1; Figure 2M and Supplementary Figure 1). ORF7b protein (mw=5179.98 Da), rich in leucine (25.58%) and phenylalanine (13.95%) is with a pI of 4.17, an instability index of 50.96, an aliphatic index of 156.51, and a grand average of hydropathicity of 1.449 (Supplementary Figure 3H; Supplementary Table 3).
No pocket was detected in the structure of ORF7b. A single tunnel with a throughput value of 0.89 was studied. The estimated high throughput tunnel 1 (blue) in ORF7b protein is with a bottleneck radius of 1.2 Å, length of 3.3 Å, distance to surface of 3.3 Å, curvature of 1.0, throughput of 0.89, and number of residues 4 (Figure 2M).
ORF8 protein: ORF8 protein has 2 sheets, 3 beta hairpins, 2 beta bulges, 8 strands, 2 helices, 16 beta turns, 4 gamma turns, and 3 disulphides (Table 1; Figures 2N and S1). ORF8 protein (mw=13830.33 Da) is rich in valine (9.92%), leucine (8.26%), and isoleucine (8.26%). ORF8 protein has presented a pI of 5.42, an instability index of 45.79, an Aliphatic index of 97.36, and a grand average of hydropathicity of 0.219 (Supplementary Figure 3I; Supplementary Table 3).
Of the six possible pockets of ORF8, the pocket with relevance score (100%), volume 401 Å3, and estimate druggability 0.75 was considered for tunnel analysis. The estimated high throughput tunnel1 (blue) in ORF 8 protein is with a bottleneck radius of 2.1 Å, length of 2.0 Å, distance to surface of 2.0 Å, curvature of 1.0, throughput of 0.92, and number of residues 11 (Figure 2N).
ORF10 protein: Structure-wise ORF10 showed 1 sheet, 1 beta alpha beta unit, 2 strands, 1 helix, and 2 beta turns (Table 1; Figure 2O and Supplementary Figure 1). The ORF10 protein (mw=4449.01 Da) is rich in asparagine (13.16%), leucine (10.53%), phenylalanine (10.53%) as well and carries pI 7.93, instability index 16.06, Aliphatic index 107.63, and grand average of hydropathicity 0.637 (Supplementary Figure 3J; Supplementary Table 3).
A single pocket with a relevance score of 100%, volume of 419 Å3 and poor estimated druggability of 0.27 was observed in ORF10 protein. The pocket showed a single tunnel (blue) with a bottleneck radius of 1.5 Å, length of 1.0 Å, distance to surface of 1.0 Å, curvature of 1.0, throughput of 0.89, and number of residues 6 (Figure 2O).
Proteomics profiles of SARS-CoV-2 proteins
Proteomics profiles of the proteins of known Experimental structures
The structure of the 3C-like proteinase presented 2 sheets, 7 beta hairpins, 7 beta bulges, 13 strands, 8 helices, 9 helix-helix interactions, 28 beta turns, and 2 gamma turns (Supplementary Figure 2) with a pI of 5.95 as well an instability index of 27.65, an aliphatic index of 82.12, and a grand average of hydropathicity of -0.019 (Supplementary Table 3).
NSP7 is predominantly an alpha helical structure with 3 helices, 7 helix-helix interactions, and 3 beta turns (PDB ID 7BV1_C, Supplementary Figure 2). NSP7 has a pI of 5.18, an instability index of 51.97, an Aliphatic index of 117.35, and a grand average of hydropathicity of 0.199 (Supplementary Table 3). NSP7 may have the function of activate RNA-synthesizing activity and form a hexadecamer with nsp8 that may participate in viral replication by acting as a primase (Xiao et al., 2012).
NSP8 has 2 sheets, 2 beta hairpins, 1 beta bulge, 5 strands, 5 helices, 6 helix-helix interactions, 13 beta turns, and 1 gamma turn (PDB ID 7BV1_B, (Supplementary Figure 2) carries a pI value of 6.58, an instability index of 37.78, an aliphatic index of 88.33, and a grand average of hydropathicity of -0.192 (Supplementary Table 3). It forms a hexa-decamer with nsp7 that may participate in viral replication by acting as a primase.
Nsp9 has a single helix with 2 sheets, 5 beta hairpins, 4 beta bulges, 7 strands, 11 beta turns (Supplementary Figure 2) showed a pI of 9.10, an instability index of 34.17, an aliphatic index of 82.92, and a grand average of hydropathicity of -0.227 (Supplementary Table 3).
The structure of NSP10 exhibited 2 sheets, 1 beta hairpin, 5 strands, 6 helices, 3 helix-helix interactions, 13 beta turns, 1 gamma turn (Supplementary Figure 2) with a pI value of 6.29, an instability index of 34.56, an aliphatic index of 61.80, and a grand average of hydropathicity of -0.068 (Supplementary Table 3). A cluster of basic residues on the protein surface suggests a nucleic acid-binding function, interacting selectively and non-covalently with an RNA molecule or a portion thereof. NSP10 contains two zincbinding motifs and forms two anti-parallel helices that are stacked against an irregular beta sheet [27].
RNA-dependent RNA polymerase (Pol/RdRp) showed 8 sheets, 8 beta hairpins, 1 psi loop, 2 beta bulges, 22 strands, 41 helices, 58 helix-helix interactions, 91 beta turns, and 16 gamma turns (Supplementary Figure 2), is associated with replication and transcription of the viral RNA genome. RNA-dependent RNA polymerase had a pI value of 6.14, an instability index of 28.32, an Aliphatic index of 78.43, and a grand average of hydropathicity of -0.224 (Supplementary Table 3).
The structure of helicase has been shown with 8 sheets, 1 beta alpha beta unit, 7 beta hairpins, 5 beta bulges, 26 strands, 19 helices, 16 helix-helix interactions, 92 beta turns, 15 gamma turns (Supplementary Figure 2) along with a pI of 8.66, an instability index of 33.31, an Aliphatic index of 84.49, and a grand average of hydropathicity of -0.096 (Supplementary Table 3).
The 3’ to 5′ exonuclease has 6 sheets, 8 beta hairpins, 3 beta bulges, 23 strands, 13 helices, 10 helix-helix interactions, and 60 beta turns (Supplementary Figure 2). It has a pI of 7.80, an instability index of 28.85, an aliphatic index of 78.96, and a grand average of hydropathicity of -0.134 (Supplementary Table 3).
EndoRNAse (NSP15) demonstrated 7 sheets, 1 beta alpha beta unit, 9 beta hairpins, 6 beta bulges, 21 strands, 10 helices, 8 helix-helix interactions, 37 beta turns, and 2 gamma turns (Supplementary Figure 2). It is with a pI of 5.06, an instability index of 36.28, an aliphatic index of 95.09, and a grand average of hydropathicity of -0.076 (Supplementary Table 3). The high-resolution crystal structure of endoribonuclease Nsp15/NendoU from SARS-CoV-2 was solved and described its catalytic domain and binding sites, which provide structural and functional evidence for developing antiviral drugs [28].
2’-O-ribose methyltransferase (NSP16) carried 3 sheets, 3 beta alpha beta units, 1 beta hairpin, 2 beta bulges, 12 strands, 12 helices, 6 helix-helix interactions,15 beta turns, and 4 gamma turns (Supplementary Figure 2) along with a pI of 7.59, an instability index of 26.11, an aliphatic index of 90.64, and a grand average of hydropathicity of -0.086 (Supplementary Table 3). The SARS-CoV RNA cap SAM-dependent (nucleoside-2’-O-)-methyltransferase (2′ -O-MTase) is a heterodimer comprising SARS-CoV nsp10 and nsp16. Nsp16 adopts a typical fold of the S-adenosylmethionine-dependent methyltransferase (SAM) family as defined initially for the catechol O-MTase,
Proteomics profiles of ORF1ab and ORF1a polyproteins
The ORF1ab polyprotein is the largest protein (7096 amino acids; 794.017kDa) of SARS-CoV-2, rich in leucine (9.41%), and valine (8.43%). The protein had a pI of 6.32, an instability index 33.31, an aliphatic index of 86.87, and a grand average of hydropathicity of −0.070 (Supplementary Figure 3K; Supplementary Table 3). It is a multifunctional protein involved in the transcription and replication of viral RNAs. It consists of proteinases responsible for the cleavage of the polyprotein. The ORF1ab polyprotein is a protein complex of 15 proteins, namely NSP1, NSP2, NSP3, NSP4, 3C-like proteinase, NSP6, NSP7, NSP8, NSP9, NSP10, RNA-dependent RNA polymerase, Helicase, 3′-to-5′ exonuclease, EndoRNAse, and 2′-O-ribose methyltransferase.
The ORF1a polyprotein (length=4405 amino acids; Molecular Weight=489.963kDa) is also rich in leucine (9.88%) and valine (8.42%). ORF1a polyprotein has a theoretical isoelectric point (pI) of 6.04, instability index of 34.92, Aliphatic index of 88.87, and Grand average of hydropathicity of −0.023 (Supplementary Figure 3L; Supplementary Table 3).
Functional annotation of SARS-CoV-2 proteome
Predicted functions of SARS-CoV-2 proteome with respective ProFunc score has been listed in Supplementary Table 4 and S5. Of the 25 proteins ORF1ab and ORF1a polyproteins are multifunctional proteins involved in the transcription and replication of viral RNAs.
Molecular phylogeny of orf1ab polyprotein
Evolutionary analysis of orf1ab polyprotein from SARSCoV- 2 was based on the Maximum Likelihood (ML) method and the JTT matrix-based model. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The ML phylogenetic tree, based on the amino acid sequence of the orf1ab of human SARS-CoV-2 (YP 009724389 and sp|P0DTD1), revealed that it has close evolutionary relatedness with new Bat coronavirus RaTG13 OX 2709072/March 2020 from China (tr|A0A6B9WIQ1) followed by Pangolin coronavirus sequenced in April 2020 (tr|A0A6G6A323 and tr|A0A6G6A2Q6) as they formed a distinct clade with a boot strap support 100% (Figure 3). Furthermore, the bat coronavirus sequenced in 2004-2005 (sp|P0C6V9, sp|P0C6W2, and sp|P0C6W6) along with Civet SARS CoV 007/2004 (Q3ZTF4) formed a clade with the human SARS coronavirus (sp|P0C6X7) with a boot strap value 100%, which is separate from novel SARS-CoV-2 (Figure 3).
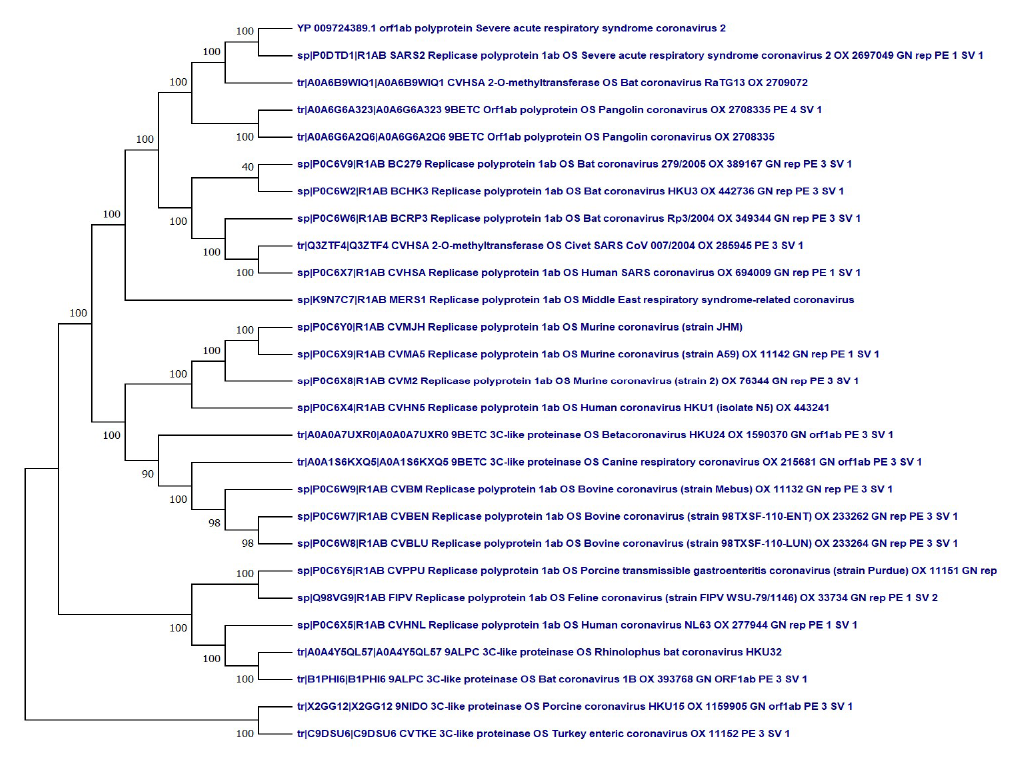
Figure 3. Evolutionary analysis of ORFab polyprotein protein of SARS-CoV-2 by Maximum Likelihood method and Le-Gascuel_model.
The tree with the highest log likelihood (-162462.82) is shown. This analysis involved 27 amino acid sequences. There was a total of 7593positions in the final dataset. Evolutionary analyses were conducted in MEGA X.
Discussion
The Neural Network- based analysis through QMEANDisCo, generated Z-scores (0, -4) and local quality estimates of the theoretical models along with Deep Learning-based quality assessment using ProQ3D (Supplementary Figure 1) are within the range of a good quality 3D structure (Table 1; Supplementary Figure 4). The TM-score indicates the difference between two structures by a score between (0,1). ProQ3D TM score of the predicted structures ranged between 0.21—0.67 (Table 1). When the TM-score <0.17, the P-value is close to 1, which means that any protein structures or computer models at this level of similarity is indistinguishable from random structure pairs. Generally, scores below 0.20 corresponds to unrelated proteins whereas structures with a score higher than 0.5 assume roughly the same fold [29]. The Deep Learning based ProQ3D and Neural Network based QMEANDisCo are advanced model quality assessments tools used in the present study from the experience of CASP13 success stories to verify the predicted 3D structures [14].
PROCHECK verification also supports that residues in the most favored regions in the Ramachandran plot in most of the models are in the range of good quality models (Table 1). The GMQE of the final theoretical models indicates that all the theoretical structures have global scores in the range of valid 3D structures (Table 1). ProQ LGscore of >3 in NSP1, surface glycoprotein, Membrane glycoprotein, NSP2, NSP3, NSP4, NSP6 indicates that the models are of good quality. Correct ProQ LGscore > 1.5 and ProQ MaxSub > 0.1 have been obtained for all the theoretical models. The computationally predicted three-dimensional structure of protein molecules has demonstrated its usefulness in many areas of biomedicine, ranging from approximate family assignments to precise drug screening. The quality of the model is directly linked to the identity between the template and target sequences, as a rule that models built over 50% sequence similarities are accurate enough for drug discovery applications, those between 25 and 50% identities can be helpful in designing mutagenesis experiments and those between 10 and 25% are tentative at superlative [30].
The instability index value of SARS-COV-2 proteins ranged between 16.06 (ORF10 protein) and 51.97 (NSP7), which classifies the ORF10 protein as the most stable and NSP7 as the most unstable protein. A protein whose instability index is smaller than 40 is predicted as stable, a value above 40 predicts that the protein may be unstable. The proteins namely ORF7a protein (48.66), ORF7b protein (50.96), NSP7 (51.97), ORF 8 protein (45.79), and nucleocapsid phosphoprotein (55.09) are unstable as per the instability index. The rest of the proteins showed stability as per the instability index (Supplementary Table 3).
The aliphatic index of a protein is the relative volume occupied by aliphatic side chains (alanine, valine, isoleucine, and leucine). It may be regarded as a positive factor for the increase of thermostability of globular proteins. The aliphatic index of SARS-COV2 ranged between 52.53 (Nucleocapsid phosphoprotein) and 156.51 (ORF 7b protein), which indicates most thermostability of the ORF 7b protein (Supplementary Table 3). The Aliphatic index indicated that SARS-COV2 proteins are thermally stable as well as they contain high amount of hydrophobic amino acids.
GRAVY (grand average of hydropathicity) index indicates the solubility of the proteins: positive GRAVY (hydrophobic), negative GRAVY (hydrophilic). The grand average hydropathicity (GRAVY) values of SARS-COV2 NSP4 (0.343), NSP6 (0.790), NSP7 (0.199), ORF3a protein (0.275), Envelope protein (1.128), membrane glycoprotein (0.446), ORF6 protein (0.233), ORF7a protein (0.318), ORF 7b protein (1.449), ORF 8 protein (0.219), and ORF10 protein (0.637) indicated that these proteins were hydrophobic in nature. All other proteins were hydrophilic (Supplementary Table 3).
The high leucine and valine content in E-protein, surface glycoprotein, ORF1ab polyprotein and ORF1a polyprotein and high leucine and isoleucine content in M-protein, ORF6 protein and ORF7b protein indicates their high structural stability (Supplementary Figure 3). Moreover, the high leucine, valine and isoleucine in ORF3a protein and ORF8 protein indicates their more structural stability than other proteins of SARS-CoV-2. The side chains of isoleucine, leucine, and valine residues often form large hydrophobic clusters that define cores of stability in high-energy states of proteins [31]. Nucleocapsid phosphoprotein is rich in glycine. Glycine is the most flexible residue, with the highest entropy of the distribution of dihedral angles (Supplementary Figure 3). Because the side chain in Glycine is absent, it has high conformational variability even in the Pro-Gly- Pro tripeptide. ORF10 protein is rich in asparagine, which is also conformationally flexible residues with glp (iXj) < 0. A low or negative value of glp (iXj) indicates that the amino acid type X has higher conformational variability than average [32].
The presence of high leucine content in ORF7a protein, ORF7b protein, E-protein, surface glycoprotein, ORF1ab polyprotein, ORF1a polyprotein, M-protein, ORF6 protein, ORF3a protein and ORF8 protein is the indication of their important roles in various protein-protein interaction processes (Supplementary Figure 3). The strong helicalforming power of leucine, as demonstrated experimentally in synthetic co-polypeptides and its high occurrence in the inner helical cores of proteins, suggests that it could have a major role as nucleation centers in the folding and evolution of large protein molecules [33].
In favorable cases, comparing 3D structures may reveal biologically interesting similarities that are not detectable by comparing sequences. A heuristic structural comparison of the newly predicted structure of the present study were made for matches against the existing full PDB entries using PDBeFold and DALI servers (Supplementary Table 6). NSP1 binds to the 40S ribosomal subunit and inhibits translation, and it also induces a templatedependent endonucleolytic cleavage of host mRNAs [34]. A comparison predicted structure of NSP1 with 18 PDB entries at a Z-score above 2.0 showed PDB IDs 2hsx-A and 2gdt-A (both are NMR structure of the NSP1 from the SARS coronavirus) highest Z-score of 22.9 and 85% of structural identity (Supplementary Table 6). Structurally, NSP1 consists of a mixed parallel/antiparallel 6-stranded beta barrel with an alpha helix covering one end of the barrel and another helix alongside the barrel [35]. NSP1 also suppresses the host innate immune functions by inhibiting type I interferon expression and host antiviral signaling pathways [35].
The SARS-CoV-2 spike glycoprotein forms complex with ACE2 and sodium-dependent neutral amino acid transporter. SARS-CoV-2 infects ciliated bronchial epithelial cells and type-II pneumocytes where an envelope-anchored spike protein binds to a host receptor angiotensin-converting enzyme 2 (ACE2) [36-37]. A comparison of 1588 matched protein structures of predicted structure of the present study with a Z-score of above 2 showed that it has high structural similarity with PDB IDs 6z97-A,6vsb-A,6zp5-A,6zow-A (the structure of the prefusion SARS-COV-2 spike glycoprotein) with a z-score of 41.3 and 93-97% of structural identity at RMSD 5.1-6.8 (Supplementary Table 6). The N-terminal S1 subunit that specifically recognizes its receptor in humans [36]. The binding efficiency of RBD on ACE2 can be enhanced by the presence of specific amino acids at the 442, 472, 479, 480, and 487 positions. Gln493 and Asn501 residues in the RBM provide possible interactions with ACE2. SARS-CoV-2 has acquired some capacity for human cell infection and human-to-human transmission by super binding affinity of Gln493 and Asn501 residues in RBM with ACE2 [38].
The envelope proteins (E proteins) are well conserved among Coronavirus strains. They are small, integral membrane proteins involved in several aspects of the virus’ life cycle, such as assembly, budding, envelope formation, and pathogenesis [39]. Of the 1626 structures matched with the predicted structure of the present study at a Z-score of above 2, the PDB IDs with closest structural fold are 5wkv, 5wkx, 5wku, 5wky, 6oqu (acid-sensing ion channel 1, 6hra (potassium-transporting ATPase potassiumbinding subunit) showed high structural identity with a z-score range of 5.0-5.6 and 9-13% of structural identity at a RMSD range of 2.5-3.3, indicating structural uniqueness of SARS-CoV-2 envelope protein (Supplementary Table 6). The E protein acts as a viroporin by oligomerizing after insertion in host membranes to create a hydrophilic pore that allows ion transport [40]. E protein from SARSCoV- 2 acts as a viroporin and self-assembles in host membranes forming ion channels. Envelope protein plays a central role in virus morphogenesis and assembly. It also induces apoptosis and IL-1β overproduction in the host cells, leading to pathogenesis. The envelope protein of SARS-CoV-2 is evolutionarily conserved with higher gene expression efficiency in the hosts [41].
The membrane (M) protein is the most abundant structural protein and defines the shape of the viral envelope. It is also regarded as the central organizer of coronavirus assembly, interacting with all other major coronaviral structural proteins. M proteins play a critical role in protein-protein interactions (as well as protein- RNA interactions) because virus-like particle (VLP) formation in many CoVs requires only the M and envelope (E) proteins for efficient virion [42]. In a comparison of 760 matched protein structures matched with the predicted structure of the present study at a Z-score above 2, PDB IDs 3ois (cysteine protease), 3pbh (pro-cathepsin B), 7pck-A (procathepsin K) showed match with a z-score range of 13.9-28.7 and 9-10% of identity at a RMSD range of 1.1-2.6 (Supplementary Table 6).
The interaction of spike (S) with M is necessary for the retention of S in the ER-Golgi intermediate compartment (ERGIC)/Golgi complex and its incorporation into new virions, but dispensable for the assembly process. Binding of M to nucleocapsid (N) proteins stabilizes the nucleocapsid (N protein-RNA complex) as well as the internal core of virions, and ultimately promotes completion of viral assembly. Together, M and E proteins make up the viral envelope, and their interaction is sufficient for the production and release of virus-like particles (VLPs) [39].
Protein 7a (SARS coronavirus X4 like protein) (Pfam: PF08779 SARS_X4) is a unique type I transmembrane protein [43]. Of the 4529 matched neighbors matched with the predicted structure of the present study at a Z-score above 2, the PDB IDs with closest structural fold are 1xak-A (88% identity), 6jmx-A, 3tcx, 1z7z-I, 1iam-A (SARS orf7a accessory protein, intercellular adhesion molecule 1, human coxsackievirus A21) with a z-score range of 5.6- 11.5 and 15-88% of structural identity at a RMSD range of 0.5-1.9 (Supplementary Table 6). It has been suggested that it binds to integrin I domains [44]. It contains a motif that has been demonstrated to mediate COPII-dependent transport out of the endoplasmic reticulum, and the protein is targeted to the Golgi apparatus (InterPro IPR01488) [45].
Nucleocapsid phosphoprotein (N proteins) form dimers, which are asymmetrically arranged into octamers via their N2b domains. The protein is a cellular component of the viral nucleocapsid (GO: 0019013). Of the 57 matched PDB structures with the predicted structure of the present study at a Z-score above 2, the PDB IDs with closest structural fold are 6g13, 7ce0, 7c22 (nucleoprotein), 6wji, 6wzq (SARSCOV- 2 nucleocapsid protein) with a z-score range of 8.9- 9.5 and 93-96% of structural identity at a RMSD of 2.7-2.8 (Supplementary Table 6). Coronavirus (CoV) nucleocapsid (N) proteins have 3 highly conserved domains. N-terminal domain (NTD) (N1b), C-terminal domain (CTD) (N2b), and N3 region. The helical nucleocapsid interacts with spike, envelope, and membrane proteins to form the assembled virion [46]. The production of gRNA in the presence of N oligomers may promote the formation of ribonucleoprotein complexes, and the newly transcribed sgmRNA would guarantee sufficient synthesis of structural proteins [47,48].
Non-structural protein 2, also known as nsp2, is an RNA-binding protein that accumulates in cytoplasmic inclusions (viroplasms). Nsp2 is involved in coronavirus (CoVs) genome replication. In a comparison of 821 protein structures matched with the predicted structure of the present study at a Z-score above 2, PDB IDs 6tqp-A, 1fyz-F, 3dxj-D (16L protein, methane monooxygenase component A, DNA-directed RNA polymerase subunit alpha) showed match with a z-score range of 3.5-4.8 and 10-13% of identity at a RMSD range of 4.9-5.4 (Supplementary Table 6).
The multi-domain Non-structural protein 3 (Nsp3) is the largest protein encoded by the coronaviruses (CoV) genome, with an average molecular mass of about 200 kD. Of the 402 neighbors matched with the predicted structure of the present study at a Z-score above 2, the PDB IDs with closest structural fold are 7cmd, 5y3q-A, 7cjd-A, 7cjd-D, 6wrh-A, 7jn2-A, 6wx4-D, 7jit-A, 7jiw-A (replicase polyprotein 1a, replicase polyprotein 1ab, papain-like protease) with a z-score range of 36-37.1 and 82-97% of structural identity at a RMSD range of 0.9-1.1 (Supplementary Table 6). It has been shown that N proteins interact with nonstructural protein 3 (NSP3) and are thus recruited to the replication-transcription complexes (RTCs). The N protein may be important for this interaction. The direct association of N protein with RTCs is a critical step for MHV infection [48]. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three-domain structure for the nucleocapsid protein [49].
NSP4 participates in the assembly of virally induced cytoplasmic double-membrane vesicles necessary for viral replication. This C-terminal domain (InterPro entry IPR032505) is predominantly alpha-helical, which may be involved in protein-protein interactions [50]. Of the 2763 structures matched with the predicted structure of the present study at a Z-score of above 2, the PDB IDs with closest structural fold are 3gzf, 3vc8, 3vcb (replicase polyprotein 1ab, RNA-directed RNA polymerase) showed high structural identity with a z-score range of 12.9-14.4 and 38-60% of structural identity at a RMSD range of 1.1- 3.1 (Supplementary Table 6). Although coexistence of nsp3 and nsp4 is known to cause membrane rearrangement, the mechanisms underlying their interactions remain unclear.
The SARS CoV-2 nsp6 proteins generate omegasome and autophagosome formation from the endoplasmic reticulum. Of the 1959 structure matched with the predicted structure of the present study at a Z-score of above 2, the PDB IDs with closest structural neighbors 6qxa-A (K+-stimulated pyrophosphate-energized sodium pump, 5gpj (pyrophosphate-energized vacuolar membrane proton) showed high structural identity with a z-score of 5.5 and 11% of structural identity at a RMSD range of 3.8-4.3 (Supplementary Table 6). NSP6 can increase cellular gene synthesis and may induce apoptosis through c-Jun N-terminal kinase, and Caspase-3 mediated stress [51] could modulate host antiviral responses by inhibiting the synthesis and signaling of interferon-beta (IFN-beta) via two complementary pathways.
Protein 3a encoded by Orf3/3a, also known as X1, which forms homotetrameric potassium, sodium, or calcium sensitive ion channels (viroporin) and may modulate virus release. It has been shown to upregulate the expression of fibrinogen subunits FGA, FGB, and FGG in host lung epithelial cells [52]. Of the 4224 structures matched with the predicted structure of the present study at a Z-score above 2, PDB ID 6bbh and 6bbg (calcium release-activated calcium channel protein 1) had highest z-score of 7.4 and 9% of structural identity at RMSD 5.1 (Supplementary Table 6). The orf3a structure matched with other PDB entries at a Z-score above 2, which calcium releaseactivated calcium channel protein, which indicates that the protein may have key role as a calcium channel protein. SARS-CoV ORF3a is a potent activator of pro-apoptosis. The expression of ORF3a induces NF-kappa B activation and upregulates fibrinogen secretion with consequent high cytokine production [52,53].
SARS-CoV ORF6 protein is localized to the endoplasmic reticulum (ER)/Golgi membrane in infected cells, where it binds to and disrupts nuclear import complex formation by tethering karyopherin alpha 2 and karyopherin beta 1 to the membrane. Of the 3038 PDB structures matched with the predicted structure of the present study at a Z-score above 2, the PDB IDs with close structural fold are 6nc8-A (lipid II flippase), 6lyp-G (mechanosensitive ion channel protein 1), 6hay-A (probable global transcription activator SNF2L2), with a z-score range of 4.5-5.5 and 11-12% of structural identity at a RMSD of 2.4-3.5 (Supplementary Table 6) Retention of import factors at the ER/Golgi membrane leads to prevention of STAT1 nuclear translocation in response to interferon signaling, thus blocking the expression of interferon-stimulated genes (ISGs) that display multiple antiviral activities [54].
ORF7b protein consists of an N-terminal, a C-terminal, and a transmembrane domain; the latter is essential to retain the protein in the Golgi compartment [55]. Despite being named as “non-structural”, it has been reported to be a structural component of SARS-CoV virions and an integral membrane protein [55]. Of the 2413 structures matched with the predicted structure of the present study at a Z-score above 2, the PDB IDs with closest structural fold are 4h9s (Histone H3.3) 6j6n (pre-mRNA-splicing factor 8), 6rqf (cytochrome b6) with a z-score range of 3.5-3.6 and 11-16% of structural identity at a RMSD of 1.4-2.3, indicating structural uniqueness of SARS-CoV-2 orf7b protein (Supplementary Table 6). Crystallographic structure (PDB ID: 6M71) and homology models revealed that RNA-directed 5’-3’ RNA polymerase activity has a unique N-terminal β-hairpin at its N-terminal [56].
The ORF8 protein of sars-cov-2 mediates immune evasion through potently downregulating MHC-I (Zhang et al., 2020). Of the 14668 matched structures with the predicted structure of the present study at a Z-score above 2, the PDB IDs with closest structural fold are 7jtl and 7jx6-A (SARS CoV-2 accessory protein NS8) with a z-score range of 15.3-17.4 and 95% of structural identity at a RMSD of 1.5 (Supplementary Table 6). All other entries showed identity below 13% indicating structural uniqueness of SARS-CoV-2 orf8 protein. ORF8 was suggested as one of the relevant genes in the study of human adaptation of the virus [57].
The protein SARS-CoV-2 ORF10 has the highest number of immunogenic epitopes of all putative ORF proteins, therefore making it a potential target for vaccine development [58]. In a comparison of 677 structures matched with the predicted structure of the present study at a Z-score above 2, PDB IDs 3w9s (SIGNALING PROTEIN), 1zgz-B (TORCAD operon transcriptional regulatory protein), 3nnn-A (DNA-binding response regulator D), showed matches with a z-score range of 3.8-4.0 and 11-17% of identity at a RMSD range of 2.6- 2.8 (Supplementary Table 6). SARS-CoV-2 ORF10 is a promising pharmaceutical target and a protein which should be monitored for changes which correlate to change pathogenesis and clinical course of COVID-19 infection.
NSP8 alone as a monomer structure may not be biologically relevant as it forms a hexadecameric supercomplex with nsp7. Nsp7-nsp8 hexadecamer may possibly confer processivity to the polymerase, may be by binding to dsRNA or by producing primers utilized by the latter. Experimental evidence for SARS-CoV that nsp7 and nsp8 activate and confer processivity to the RNA-synthesizing activity of Polymerase [59].
Nsp10 plays a pivotal role in viral transcription by stimulating nsp14 3’-5’ exoribonuclease activity. Nsp10 plays a pivotal role in viral transcription by stimulating nsp16 2’-O-ribose methyltransferase activity. Spike protein S1 binds to human ACE2, initiating infection. CoV attaches to the target cells with the help of spike protein–host cell protein interaction (angiotensin converting enzyme-2 (ACE-2) interaction in SARS-CoV [60], and dipeptidyl peptidase-4 [DPP-4] in MERS-CoV [61]. After receptor recognition, the virus genome with its nucleocapsid is released into the cytoplasm of host cells.
The 3C-like polyprotein is a target of drug cinanserin, acting most likely via inhibition of the 3C-like proteinase, which strongly reduces virus replication identified by deploying both a homology model and the crystallographic structure of the binding pocket of the enzyme [62]. NSP9 (PF08710) is a single-stranded RNA-binding viral protein likely involved in RNA synthesis [63], and its structure comprises a single beta barrel [64]. NSP7 and NSP8 activate and confer processivity to the RNA-synthesizing activity of Pol [59].
The 3C-like polyprotein is a target of drug cinanserin, acting most likely via inhibition of the 3C-like proteinase, which strongly reduces virus replication identified by deploying both a homology model and the crystallographic structure of the binding pocket of the enzyme [62]. NSP9 (PF08710) is a single-stranded RNA-binding viral protein likely involved in RNA synthesis [63], and its structure comprises a single beta barrel [64]. NSP7 and NSP8 activate and confer processivity to the RNA-synthesizing activity of Pol [59].
Tunnels are access paths connecting the interior of molecular systems with the surrounding environment. The presence of tunnels in proteins influences their reactivity, as they determine the nature and intensity of the interaction that these proteins can take part in [65]. Tunnel and ligand-binding pocket analysis in the newly predicted structures of the present study had estimated pockets with high druggability scores in envelope glycoprotein (0.97), membrane glycoprotein (0.87), NSP6 (0.79), ORF7a (0.79), ORF8 (0.75), ORF3a (0.72), and NSP4 (0.70), indicating the ability to bind drug-like molecules with high affinity, which is of major interest in the target identification phase of drugs. Multiple pockets with high druggability pockets have been detected in the nucleocapsid phosphoprotein (03), NSP3 (10), membrane glycoprotein (08), NSP4 (10), and NSP6 (10), NSP1 (08), and ORF7a (04).Tunnel analysis of the high-relevance sore pockets has estimated the presence of multiple tunnels in NSP4 (seven), nucleocapsid phosphoprotein (seven), NSP3 (five), membrane glycoprotein (five), ORF6 protein (four), NSP1 (three), NSP6 (three), and envelop protein (03).The presence of multiple tunnels in these proteins may take a key role in a large number of transport pathways for small ligands influencing their reactivity. However, pockets with poor druggability scores were observed in surface glycoprotein (0.10), NSP2 (0.31), ORF6 (0.03), and ORF10 (0.27). No active pocket was observed in ORF7b. Pocket druggability investigations represent a key step in compound clinical progression projects. It has been experimentally demonstrated that the tunnels and their properties can define many important protein characteristics like substrate specificity, enantioselectivity, stability, and activity [66]. After verification, the coordinate files were successfully deposited to ModelArchive (https://www.modelarchive.org).The details of the verified structures along with verification report have been deposited to Modelarchive are available to download along with the structures (https://www.modelarchive.org ; Table 1; Annexure S1).
Our study on whole-genome phylogenetic tree strongly supports the protein phylogeny based on orf1ab polyprotein, indicating that the close evolutionary SARS-CoV-2 has very closely evolutionarily related to newly sequenced Bat coronavirus RaTG13 genome /March 2020 from China (MN996532) followed by the pangolin coronavirus genome (MT040333, MT040335, MT072864) [6]. The present study is supported by our previous study as both strongly suggests that like the human host the coronavirus had undergone rapid evolution in bats and pangolin as an amplifying host (Figure 3).
Earlier research claimed that cross-species transmission of zoonotic coronaviruses (CoVs) can result in disease outbreaks [67]. The close phylogenetic relationship to RaTG13 provides evidence that 2019-nCoV may have originated in bats [41]. Molecular analysis supported bats as natural hosts for SARS-CoV, but palm civets (Paguma larvata) had a critical role in the transmission to humans [68,69]. Bats are implicated in SARSCoV-2 origin. A very similar SARS-CoV-2 strain (RaTG13 CoV) was detected in Rhinolophus affinis bat with 96% genome similarity compared with SARS-CoV-2 genome sequence. The comparison study from bat and pangolin explained that BetaCoV/bat/Yunnan/RaTG13/2013 virus was more similar to the SARS-CoV-2 virus than the coronavirus obtained from the two pangolin samples (SRR10168377 and SRR10168378) [37]. This indicates that the human SARS-CoV-2 virus, which is responsible for the recent outbreak of COVID-19, did not come directly from pangolins.
Naturally occurring proteins are the nanoscale machines that carry out nearly all the essential functions in living things. Proteins can be classified as nanostructures because the size is around 1–100 nm. The use of proteins in nanotechnology is a largely unexplored area. However, due to their complex structure, proteins offer many possibilities to develop effective nanocarriers to counter the conventional limitations of antiviral and biological therapeutics [70]. Nanocarriers have potential to design risk-free and effective immunization strategies for SARSCoV-2 vaccine candidates such as protein constructs and nucleic acids[70].Nanotechnology benefits nextgeneration vaccine design since nanomaterials are ideal for antigen delivery, as adjuvants, and as mimics of viral structures. Subunit vaccines can also take the form of protein nanoparticles or virus-like particles (VLPs). Developing peptide epitope vaccine strategies targeting the SARS-CoV-2 S protein may yield a safer vaccine [71]. Using genetic information and protein structure modeling, several nanotherapeutic strategies based on drug repurposing may be projected for the immediate treatment of infected patients of COVID-19 [70].
Conclusion
The RNA genome of SARS-CoV-2 has 29.9 kb nucleotides, encoding 29 proteins, although one may not be expressed. Studying these different components of the virus as well as how they interact with human cells have already yielded some clues, but much remains to be explored. The present study reported the theoretical modeling of 15 proteins. In silico sequence-based and structure-based functional characterization of the full SARS-CoV-2 proteome based on the NCBI reference sequence NC_045512 (29903 bp ss-RNA). The presence of a large number of tunnels in NSP1, nucleocapsid phosphoprotein, NSP3, membrane glycoprotein, ORF6 protein, NSP6, ORF3a protein, and ORF7a protein indicates their high reactivity. The theoretical structures and statistical verification reports were successfully deposited in the Model Archive. The 15 theoretical structures of novel SARS-CoV-2 would perhaps be useful for advanced computational analysis of interactions of each protein for detailed functional analysis of active sites toward structure-based drug design or to study potential advanced vaccines using nano-intervention, if at all, towards prevent epidemics and pandemics in the absence of a complete experimental structure. Phylogenetic analysis of orf1ab polyprotein revealed a close evolutionary relationship between the newly emerged human SARS CoV-2 and bat SARS-like coronavirus. The predicted protein structures may be useful in nanocarrier-based therapeutics to offers several opportunities to address the limitations of current antiviral therapy for the COVID-19 treatment.
Data Availability
(1) The resultant protein structures are deposited in ModelArchive (https://www.modelarchive.org/). The same data has been provided in a supplementary file.
(2) The supplementary file for the data generated in the project has been deposited to BioRxiv. Preprint doi: 10.1101/2020.05.23.104919 (supplementary file) and accessible at Europe PMC (PPR166822).
(3) All the above data are also included along with this manuscript.
Authors’ Contributions
P Devi and DKS were involved in planning, designing, and realizing the present work. Data acquisition and analysis were done by CB and SM. After in-depth discussion of the results with DKS, both CB and PD prepared the manuscript, and SM finalized the manuscript.
Acknowledgements
The authors are grateful to DBT-Govt. of India forsupporting Bioinformatics infrastructure (under the DBTStar College scheme) at the Post Graduate Department of Zoology, Darrang College, Tezpur, Assam. The authors are thankful to the Principal, Darrang College (Gauhati University), Tezpur (Assam), India, and Head of the Post Graduate Department of Zoology, Darrang College, and the University of Science and Technology, Meghalaya, India, for supporting the research laboratory facility.
References
2. Lai MC. Coronaviridae. Fields Virology. 2007:1305-18.
3. Lu G, Liu D. SARS-like virus in the Middle East: a truly bat-related coronavirus causing human diseases. Protein & Cell. 2012 Nov 1;3(11):803-5.
4. Chang CK, Jeyachandran S, Hu NJ, Liu CL, Lin SY, Wang YS, et al. Structure-based virtual screening and experimental validation of the discovery of inhibitors targeted towards the human coronavirus nucleocapsid protein. Molecular Biosystems. 2016;12(1):59-66.
5. Paules CI, Marston HD, Fauci AS. Coronavirus infections—more than just the common cold. Jama. 2020 Feb 25;323(8):707-8.
6. Baruah C, Devi P, Sharma DK. Sequence analysis and structure prediction of SARS-CoV-2 accessory proteins 9b and ORF14: evolutionary analysis indicates close relatedness to bat coronavirus. BioMed Research International. 2020: 7234961.
7. Floudas CA, Fung HK, McAllister SR, Mönnigmann M, Rajgaria R. Advances in protein structure prediction and de novo protein design: A review. Chemical Engineering Science. 2006 Feb 1;61(3):966-88.
8. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research. 1997 Sep 1;25(17):3389-402.
9. Pearson WR. Searching protein sequence libraries: comparison of the sensitivity and selectivity of the Smith- Waterman and FASTA algorithms. Genomics. 1991 Nov 1;11(3):635-50.
10. Webb B, Sali A. Comparative protein structure modeling using MODELLER. Current Protocols in Bioinformatics. 2016 Jun;54(1):5-6.
11. Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research. 2018 Jul 2;46(W1):W296-303.
12. Fiser A, Do RK, Šali A. Modeling of loops in protein structures. Protein Science. 2000;9(9):1753-73.
13. Eswar N, Webb B, Marti-Renom MA, Madhusudhan MS, Eramian D, Shen MY, et al. Comparative protein structure modeling using Modeller. Current Protocols in Bioinformatics. 2006 Sep;15(1):5-6.
14. Haas J, Gumienny R, Barbato A, Ackermann F, Tauriello G, Bertoni M, et al. Introducing “best single template” models as reference baseline for the Continuous Automated Model Evaluation (CAMEO). Proteins: Structure, Function, and Bioinformatics. 2019 Dec;87(12):1378-87.
15. Williams CJ, Headd JJ, Moriarty NW, Prisant MG, Videau LL, Deis LN, et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Science. 2018 Jan;27(1):293-315.
16. Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. Journal of Applied Crystallography. 1993 Apr 1;26(2):283-91.
17. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004 Oct;25(13):1605-12.
18. Hutchinson EG, Thornton JM. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Science. 1996 Feb;5(2):212-20.
19. Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In the Proteomics Protocols Handbook 2005 (pp. 571-607). Humana Press.
20. Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. InNucleic Acids Symposium Series 1999 Jan 1 (Vol. 41, No. 41, pp. 95-98). [London]: Information Retrieval Ltd., c1979-c2000.
21. Laskowski RA, Watson JD, Thornton JM. ProFunc: a server for predicting protein function from 3D structure. Nucleic Acids Research. 2005 Jul 1;33(suppl_2):W89-93.
22. Hutchinson EG, Thornton JM. PROMOTIF—a program to identify and analyze structural motifs in proteins. Protein Science. 1996 Feb;5(2):212-20.
23. Stourac J, Vavra O, Kokkonen P, Filipovic J, Pinto G, Brezovsky J, et al. Caver Web 1.0: identification of tunnels and channels in proteins and analysis of ligand transport. Nucleic Acids Research. 2019 Jul 2;47(W1):W414-22.
24. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994 Nov 11;22(22):4673-80.
25. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution. 2018 Jun 1;35(6):1547-9.
26. Le SQ, Gascuel O. An improved general amino acid replacement matrix. Molecular Biology and Evolution. 2008 Jul 1;25(7):1307-20.
27. Joseph JS, Saikatendu KS, Subramanian V, Neuman BW, Brooun A, Griffith M, et al. Crystal structure of nonstructural protein 10 from the severe acute respiratory syndrome coronavirus reveals a novel fold with two zinc-binding motifs. Journal of Virology. 2006 Aug 15;80(16):7894-901.
28. Kim Y, Jedrzejczak R, Maltseva NI, Wilamowski M, Endres M, Godzik A, et al. Crystal structure of Nsp15 endoribonuclease NendoU from SARS-CoV-2. Protein Science. 2020 Mar 3.
29. Xu J, Zhang Y. How significant is a protein structure similarity with TM-score= 0.5?. Bioinformatics. 2010 Apr 1;26(7):889-95.
30. Wong WC, Maurer-Stroh S, Eisenhaber F. Not all transmembrane helices are born equal: Towards the extension of the sequence homology concept to membrane proteins. Biology Direct. 2011 Dec 1;6(1):57.
31. Kathuria SV, Chan YH, Nobrega RP, Özen A, Matthews CR. Clusters of isoleucine, leucine, and valine side chains define cores of stability in high-energy states of globular proteins: Sequence determinants of structure and stability. Protein Science. 2016 Mar;25(3):662-75.
32. Kuznetsov IB, Rackovsky S. On the properties and sequence context of structurally ambivalent fragments in proteins. Protein Science. 2003 Nov;12(11):2420-33.
33. Chou PY, Fasman GD. Structural and functional role of leucine residues in proteins. Journal of Molecular Biology. 1973 Mar 5;74(3):263-81.
34. Kamitani W, Narayanan K, Huang C, Lokugamage K, Ikegami T, Ito N, et al. Severe acute respiratory syndrome coronavirus nsp1 protein suppresses host gene expression by promoting host mRNA degradation. Proceedings of the National Academy of Sciences. 2006 Aug 22;103(34):12885-90.
35. Almeida MS, Johnson MA, Herrmann T, Geralt M, Wüthrich K. Novel ß-barrel fold in the nuclear magnetic resonance structure of the replicase nonstructural protein 1 from the severe acute respiratory syndrome coronavirus. Journal of Virology. 2007 Apr 1;81(7):3151-61.
36. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020 May;581(7807):215- 20.
37. Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, et al. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. Journal of Medical Virology. 2020 Jun;92(6):602-11.
38. Wan Y, Shang J, Graham R, Baric RS, Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. Journal of Virology. 2020 Mar 17;94(7).
39. Schoeman D, Fielding BC. Coronavirus envelope protein: current knowledge. Virology Journal. 2019 Dec;16(1):1-22.
40. Surya W, Li Y, Verdià-Bàguena C, Aguilella VM, Torres J. MERS coronavirus envelope protein has a single transmembrane domain that forms pentameric ion channels. Virus Research. 2015 Apr 2;201:61-6.
41. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020 Mar;579(7798):270-3.
42. Ujike M, Taguchi F. Incorporation of spike and membrane glycoproteins into coronavirus virions. Viruses. 2015 Apr;7(4):1700-25.
43. Nelson CA, Pekosz A, Lee CA, Diamond MS, Fremont DH. Structure and intracellular targeting of the SARScoronavirus Orf7a accessory protein. Structure. 2005 Jan 1;13(1):75-85.
44. Hänel K, Stangler T, Stoldt M, Willbold D. Solution structure of the X4 protein coded by the SARS related coronavirus reveals an immunoglobulin like fold and suggests a binding activity to integrin I domains. Journal of Biomedical Science. 2006 May 1;13(3):281-93.
45. Pekosz A, Schaecher SR, Diamond MS, Fremont DH, Sims AC, Baric RS. Structure, expression, and intracellular localization of the SARS-CoV accessory proteins 7a and 7b. InThe Nidoviruses 2006 (pp. 115-120). Springer, Boston, MA.
46. Zumla A, Chan JF, Azhar EI, Hui DS, Yuen KY. Coronaviruses—drug discovery and therapeutic options. Nature Reviews Drug Discovery. 2016 May;15(5):327-47.
47. Wu CH, Chen PJ, Yeh SH. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host & Microbe. 2014 Oct 8;16(4):462-72.
48. Cong Y, Ulasli M, Schepers H, Mauthe M, V’kovski P, Kriegenburg F, et al. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. Journal of Virology. 2020 Jan 31;94(4).
49. Parker MM, Masters PS. Sequence comparison of the N genes of five strains of the coronavirus mouse hepatitis virus suggests a three domain structure for the nucleocapsid protein. Virology. 1990 Nov 1;179(1):463-8.
50. Manolaridis I, Wojdyla JA, Panjikar S, Snijder EJ, Gorbalenya AE, Berglind H, et al. Structure of the C-terminal domain of nsp4 from feline coronavirus. Acta Crystallographica Section D: Biological Crystallography. 2009 Aug 1;65(8):839-46.
51. Cheng W, Chen S, Li R, Chen Y, Wang M, Guo D. Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin-dependent proteosomal degradation of N-Myc (and STAT) interactor. Virologica Sinica. 2015 Apr 1;30(2):153-61.
52. Lu W, Zheng BJ, Xu K, Schwarz W, Du L, Wong CK, et al. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proceedings of the National Academy of Sciences. 2006 Aug 15;103(33):12540-5.
53. Yu CJ, Chen YC, Hsiao CH, Kuo TC, Chang SC, Lu CY, et al. Identification of a novel protein 3a from severe acute respiratory syndrome coronavirus. FEBS Letters. 2004 May 7;565(1-3):111-6.
54. Frieman M, Yount B, Heise M, Kopecky-Bromberg SA, Palese P, Baric RS. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. Journal of Virology. 2007 Sep 15;81(18):9812-24.
55. Schaecher SR, Mackenzie JM, Pekosz A. The ORF7b protein of severe acute respiratory syndrome coronavirus (SARS-CoV) is expressed in virus-infected cells and incorporated into SARS-CoV particles. Journal of Virology. 2007 Jan 15;81(2):718-31.
56. Gao Y, Yan L, Huang Y, Liu F, Zhao Y, Cao L, et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020 May 15;368(6492):779-82.
57. Law PY, Liu YM, Geng H, Kwan KH, Waye MM, Ho YY. Expression and functional characterization of the putative protein 8b of the severe acute respiratory syndrome-associated coronavirus. FEBS Letters. 2006 Jun 26;580(15):3643-8.
58. Kiyotani K, Toyoshima Y, Nemoto K, Nakamura Y. Bioinformatic prediction of potential T cell epitopes for SARS-Cov-2. Journal of Human Genetics. 2020 Jul;65(7):569-75.
59. Kirchdoerfer RN, Ward AB. Structure of the SARSCoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nature Communications. 2019 May 28;10(1):1-9.
60. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003 Nov;426(6965):450-4.
61. Mubarak A, Alturaiki W, Hemida MG. Middle east respiratory syndrome coronavirus (MERS-CoV): infection, immunological response, and vaccine development. Journal of Immunology Research. 2019 Apr 7;2019.
62. Chen L, Gui C, Luo X, Yang Q, Günther S, Scandella E, et al. Cinanserin is an inhibitor of the 3C-like proteinase of severe acute respiratory syndrome coronavirus and strongly reduces virus replication in vitro. Journal of Virology. 2005 Jun 1;79(11):7095-103.
63. Egloff MP, Ferron F, Campanacci V, Longhi S, Rancurel C, Dutartre H, et al. The severe acute respiratory syndrome-coronavirus replicative protein nsp9 is a singlestranded RNA-binding subunit unique in the RNA virus world. Proceedings of the National Academy of Sciences. 2004 Mar 16;101(11):3792-6.
64. Campanacci V, Egloff MP, Longhi S, Ferron F, Rancurel C, Salomoni A, et al. Structural genomics of the SARS coronavirus: cloning, expression, crystallization and preliminary crystallographic study of the Nsp9 protein. Acta Crystallographica Section D: Biological Crystallography. 2003 Sep 1;59(9):1628-31.
65. Jaiswal D, Vareková RS, Ionescu CM, Sehnal D, Koca J. Searching for tunnels of proteins–comparison of approaches and available software tools. Journal of Cheminformatics. 2012 Dec 1;4(S1):P60.
66. Brezovsky J, Babkova P, Degtjarik O, Fortova A, Gora A, Iermak I, et al. Engineering a de novo transport tunnel. ACS Catalysis. 2016 Nov 4;6(11):7597-610.
67. Agnihothram S, Yount BL, Donaldson EF, Huynh J, Menachery VD, Gralinski LE, et al. A mouse model for Betacoronavirus subgroup 2c using a bat coronavirus strain HKU5 variant. MBio. 2014 May 1;5(2).
68. Li W, Wong SK, Li F, Kuhn JH, Huang IC, Choe H, et al. Animal origins of the severe acute respiratory syndrome coronavirus: insight from ACE2-S-protein interactions. Journal of Virology. 2006 May 1;80(9):4211-9.
69. Wang LF, Eaton BT. Bats, Civets and the emergence of SARS. In Wildlife and emerging zoonotic diseases: the biology, circumstances and consequences of crossspecies transmission 2007 (pp. 325-344). Springer, Berlin, Heidelberg.
70. Chauhan G, Madou MJ, Kalra S, Chopra V, Ghosh D, Martinez-Chapa SO. Nanotechnology for COVID-19: therapeutics and vaccine research. ACS Nano. 2020 Jun 22;14(7):7760-82.
71. Shin MD, Shukla S, Chung YH, Beiss V, Chan SK, Ortega-Rivera OA, et al. COVID-19 vaccine development and a potential nanomaterial path forward. Nature Nanotechnology. 2020 Aug;15(8):646-55.