Abstract
Polyploid cancer cells can arise de novo in tumors or they can be induced by therapeutics that inadvertently increase the rate of cytokinetic failure. These cells portend a poor outcome in many cancers because polyploid cancer cells can undergo error prone reductive cell divisions to yield aneuploid progeny. The immune system has evolved mechanisms by which it can specifically recognize and remove polyploid cancer cells, but these appear to be tampered with malignancy so that polyploid cells can persist and fuel the development of cancer cell clones that are resistant to therapeutics and have metastatic potential. Here we review mechanisms by which polyploid cancer cells can arise, are surveilled by the immune system and therapeutic strategies that might prevent or directly attack polyploid cancer cells.
Keywords
Polyploid, Mitosis, Therapeutics, Apoptosis, Cancer, Immune surveillance
Normal Polyploid Cells
While cells with a 2n complement of chromosomes are defined as diploid, cells that possess greater than 2n are referred to as polyploid. The additional DNA content of polyploid cells may seem highly divergent, but polyploid cells exist in mammals and serve vital roles in both development and tissue homeostasis [1]. Placental syncytiotrophoblasts, for example, form the interface between the maternal blood and embryonic fluid. These cells enable gas and nutrient exchange and produce hormones that maintain pregnancy. They are multinucleate cells and are formed and maintained through fusion of the underlying diploid cytotrophoblast cells [2]. Polyploidization can alternatively occur when the genome is replicated but cells fail to undergo cytokinesis. This situation occurs with the maturation of megakaryocytes (MKs). They develop from diploid, bone marrow hematopoietic stem cells, but during maturation they become polyploid by endoreplication, the replication of DNA without cell division. This process is driven by the hormone Thrombopoietin and can result in up to 64n DNA content [3]. The final maturation step for MKs requires extrusion of nuclear content and the formation of proplatelet structures from their cytoplasm. Although still debated, the requirement for polyploidy in MK development is likely related to the requirement for large quantities of mRNA and protein that is eventually packaged into platelets for clotting and repair.
Syncytiotrophoblasts, formed through cell fusion, and MKs, through endoreplication, represent two distinct, specialized cell types where polyploidy plays a vital physiological role. However, aberrant polyploid cancer cells can also arise when the normal checks and balances on cell division are compromised in pseudodiploid cancer cells. Below, we outline how polyploid cancer cells arise and the rationale for attacking this population of cancer cells.
Polyploid Cancer Cells
Oncogenic changes that occur in cancer facilitate mitotic slippage and cytokinetic failure. This disruption facilitates aneuploidy, a numerical change in a fraction of the diploid set of chromosomes. For example, loss-of-function mutations in tumor suppressors such as BRCA2 [4], TP53 [5] and APC [6], all increase the rate of cytokinetic failure, while activating kinase mutations can impact the fidelity of mitosis. Signaling cascades converge to influence the biogenesis and function of centrosomes, the integrity of the mitotic spindle assembly checkpoint (SAC), and the completion of cytokinesis [7-9]. The SAC acts as a safeguard for the accurate segregation of chromosomes, ensuring proper attachment of kinetochores to microtubules of the mitotic spindle and optimal tension between bi-oriented sister kinetochores before the transition to anaphase (for review see [10-12]). Defects in chromosome segregation are thought to result from bypass of the SAC. Thus, prior oncogenic events can serve as a prelude to further intratumoral genetic heterogeneity by promoting aneuploidy. This fuels the emergence of more aggressive cancer cell clones over time. Single cell sequencing technologies continue to reveal enormous depth in the clonal heterogeneity found in cancer [13].
However, a prominent pathway to aneuploidy may involve a polyploid intermediate. Polyploidy differs from aneuploidy. Polyploidy is a numerical change in the whole set of chromosomes, not just a fraction. Cancer cells can undergo a viable mitosis but then fail to complete cytokinesis, resulting in the formation of a multinucleate cell. In contrast with the conventional view of polyploidy as a proliferatively arrested state, accumulated data indicate that polyploid cells can undergo reductive divisions that may be error prone, resulting in highly aneuploid progeny that are viable and proliferative [14]. Compared with diploidy, polyploidy serves as a resilient intermediate for aneuploidy because the increased DNA content buffers the loss of essential chromosomes more effectively [15].
Cells that are 4n, 8n or more are present in many tumors and the presence of polyploid cells is recognized as a poor prognostic indicator in multiple cancers [16-18]. For leukemias, in particular, it has long been recognized to portend poor outcome [19]. So, the polyploid pool of tumor cells can serve as a constant source upon which cells with variable genomic alterations can emerge, producing therapeutically resistant cells and cells with enhanced metastatic potential over time [14,15,20] (outlined in Figure 1).
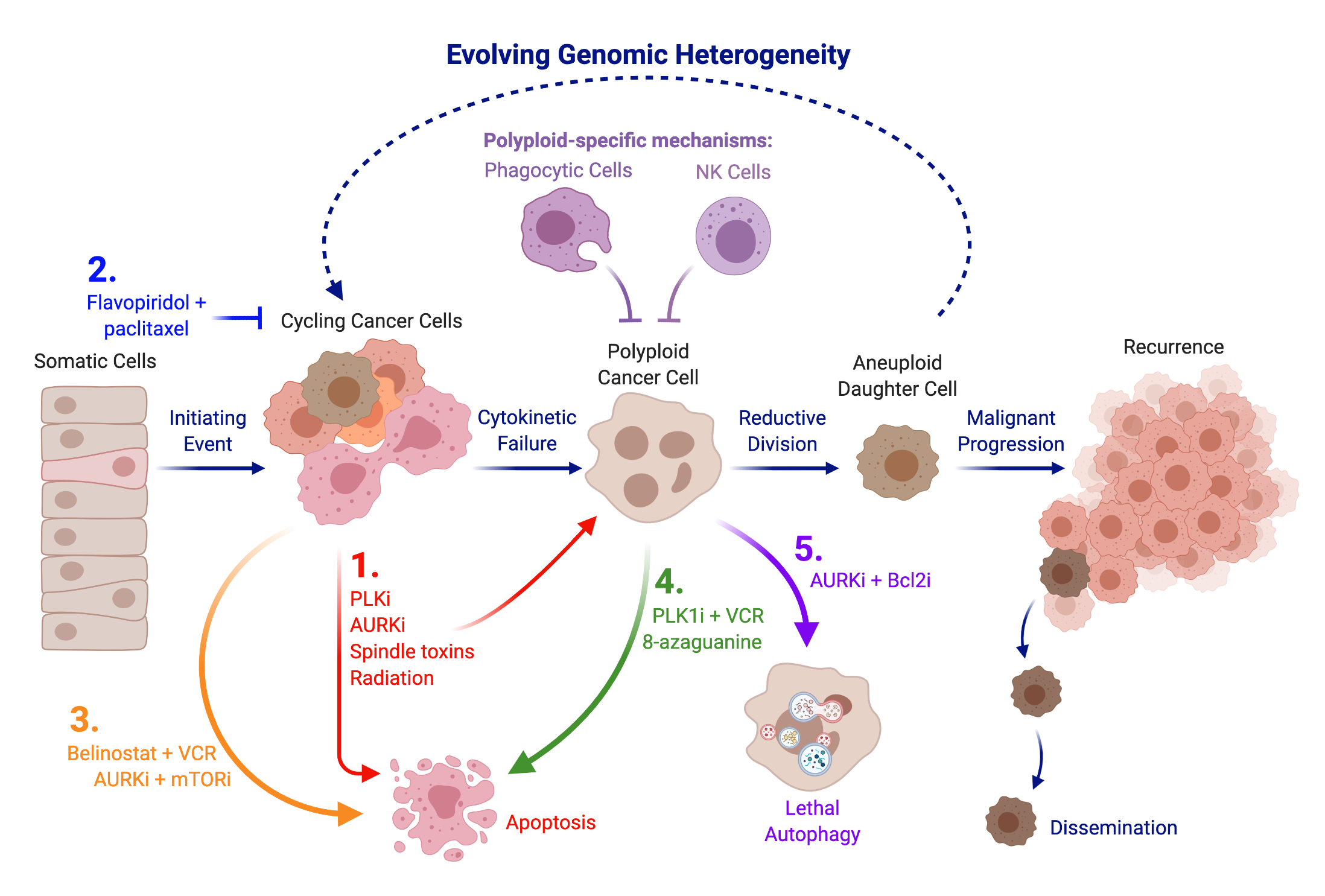
Figure 1: Prophylactic and therapeutic strategies for polyploid cells. Illustrated are steps in the maligna nt progression of cancer and potential therapeutic strategies against polyploid cancer cells. Polyploid cancer cells are a precursor to aneuploid cancer cells through error-prone reductive cellular division and are subject to immune surveillance. Polyploid cells seed the constantly evolving genomic heterogeneity within tumors, spawning clones that are drug refractory and/or have metastatic potential. Therapeutics mentioned in this review that interfere with this process are shown. 1. Therapeutics that attack mitotic cells can induce apoptosis in some cancer cells but also can provoke a subpopulation of polyploid cancer cells that escape from apoptosis (red). 2. Combining flavopiridol (CDK inhibitor) with paclitaxel induces G1 arrest that minimizes development of polyploidy (blue). 3. Combination therapies can alter the propensity for polyploidy by enhancing mitotic catastrophe-induced death of cycling cancer cells (orange). 4. Treatments can show enhanced propensity to attack newly generated polyploid cancer cells, presumably due to vulnerabilities inherent in polyploid cancer cells (green) 5. Inhibition of the interaction between Bcl2 and Beclin1 can enable lethal autophagy as a means to directly attack polyploid cancer cells (purple). Note: A lower case i denotes an inhibitor, i.e. Bcl2i is an inhibitor of Bcl2, such as a Bcl2 mimetic; VCR is vincristine.
Polyploid cells appear particularly well suited to seed metastatic recurrence. One response to polyploidy is the development of cellular senescence [21]. As quiescent cells, polyploids may be uniquely able to survive in the face of chemotherapeutics that target dividing cells. In addition, DNA damage response genes are rewired in polyploid cells, triggering single-stranded base repair and non-homologous end-joining pathways to increase DNA repair activity [22]. The emergence of polyploid cells from senescence to produce viable aneuploid progeny could contribute to tumor recurrence long after chemotherapy has ceased. Multiple lines of evidence suggest they do, in fact, emerge and produce aggressive cancer cell clones [14,20,23,24]. One not fully understood aspect of polyploid cancer cell biology is the observed enhancement in polyploids of properties that are associated with highly malignant cancers, even when these properties are not evident in the associated diploid cancer cell pool. Examples include altered expression of cell cycle regulators [22,25] and markers associated with epithelial to mesenchymal transition (EMT) and cancer stem cells [26,27]. A detailed review of the enhancement of malignant properties in polyploid cancer cells has been published [28].
Importantly, polyploid cancer cells have the ability to seed tumorigenesis, so they do have cancer stem cell-like properties. Polyploid cells isolated from ovarian cancer cell lines express higher levels of the ovarian cancer stem cell marker CD133, form spheroids in culture and tumors in immunocompromised mice [27]. Perhaps most telling regarding the dedifferentiation phenotype of these cells is that they can be selectively manipulated in cell culture to take on properties of mesenchymal lineages from adipose, cartilage and bone [27]. The propensity to undergo EMT has long been considered a factor that portends metastatic dissemination, so the inheritance of developmental plasticity may be an important characteristic that daughter cells inherit from a polyploid precursor.
Such inheritance might be epigenetic. There is evidence, at least in p53 positive cancer, that epigenetic changes in polyploid cells enable silencing of p53 transciptional targets that activate apoptosis and cell cycle arrest. For example, the the DNA methylation inhibitor 5-aza-2- deoxycytidine (5-AzadC) can restore expression of the p53 target and cyclin-dependent kinase inhibitor, p21CIP1, and also restore polyploid cancer cell sensitivity to TNFα [22]. The ability of epigentic alterations to be passed on to the aneuploid progeny of polyploids has not been sufficiently explored and may contribute to the emergence of drug resistance and metastasis.
Immunosurveillance
In immunocompetent mice, tumors that eventually emerge following polyploid cell engraftment are comprised mainly of pseudodiploid cancer cells, so they arise from the progeny of a reductive cell division [29]. The ability of the immune system to specifically detect polyploid cells could be a mechanism that necessitates this reduction in ploidy.
Mechanisms by which the immune system eliminates polyploid cancer cells arise through stress signaling. The protein calreticulin becomes relocalized to the plasma membrane of polyploid cancer cells, where it acts as a ligand for LDL-receptor-related protein (LRP) (also known as CD91) on the surface of phagocytic cells [30]. To act as an “eat me” signal, translocation must occur from the endoplasmic reticulum (ER) where calreticulin normally functions as a molecular chaperone [31]. Strong evidence that calreticulin is central to polyploid cell immunosurveillance comes from experiments demonstrating calreticulin exposure on the cell surface of polyploid cells does not limit tumorigenesis in immunodeficient mice, but does limit tumorigenesis in mice with an intact immune system [29,32]. Constitutive ER stress in polyploid cells directs calreticulin to the cell surface as manipulations that alleviate ER stress also reduce calreticulin transport to the cell surface and immunogenicity [29].
Polyploid cells are also subject to heightened immunosurveillance by Natural Killer (NK) cells. Hyperploidy-inducing chemotherapeutics induce cell surface expression of ligands for the NK cell-activating receptors NKG2D and DNAM-1 [33]. Again, the ER stress response plays a role. The NKG2D ligand, MICA is upregulated on the surface of both HCT-116 colon cancer cells and K-562 myelogenous leukemia cells by ER stress and this triggers the cytolytic activity of NKs [33]. Polyploidy may therefore prime anti-tumor immunity. Despite this constant immunosurveillance, cancers are often diagnosed at an advanced stage with aneuploidy, dissemination and innate ability to avoid immune effector cells. Despite the advent of immune checkpoint modulating antibodies, restoring the ability of the immune system to identify and attack cancer cells is still a major clinical challenge.
Chemotherapy-Induced Polyploidy
Added complexity stems from the fact that therapies that attack dividing cells can inadvertently enhance the development of polyploidy. For example, drugs that disrupt the mitotic spindle induce prolonged mitotic arrest leading to mitotic catastrophe-induced apoptosis. However, sporadic cells escape, perhaps by falling beneath the threshold of inductive cues required for apoptosis. These cells can alternatively fail to complete cytokinesis, becoming polyploid. Successive rounds of replication with failed cytokinesis can yield cells with even greater than 4n ploidy and while apoptotic signaling is still viable in tetraploid cancer cells induced by nocodazole [34], tetraploid cell lines are more resistant to radiation and DNA damage-induced death than their diploid counterparts [35]. This suggests polyploidization goes hand-in-hand with dampened ability to activate intrinsic apoptotic signaling. Distinct from apoptosis, enhanced autophagic flux in polyploid cancer cells has been found to either promote or suppress their long-term survival in a context-dependent manner [36,37]. The persistence of polyploid cancer cells is likely enabled by the convergence of multiple escape mechanisms.
Several classes of cancer therapy induce a polyploid cell population, including the clinically used taxanes such as docetaxel [24,38] and paclitaxel, [39], DNA damaging agents such as doxorubicin [40], radiation [14,41] and oncoprotein-targeting compounds [7,42]. This also includes targeted kinase inhibitory drugs that directly attack the mitotic machinery, such as Aurora kinase inhibitors [37,43] and Polo-like kinase inhibitors [44,45]. If cycles of polyploidization followed by reductive cell division fuel the progressive generation of aneuploidy, then a therapeutic that targets polyploid cancer cells may be a means to short circuit this cycling, attack the evolvability of the cancer genome and enhance the overall effectiveness of many currently used therapies.
Preventing the Development of Polyploid Cancer Cells
In theory, reducing conversion of diploid cancer cells to polyploid cells could be a strategy to limit evolvability. Several lines of evidence suggest combination therapies that work toward this goal. One combination therapy that effectively targets polyploid cell formation, at least in cultured diffuse large B cell lymphoma (DLBCL) cell lines, is the combined use of the histone deacetylase complex (HDAC) inhibitor, Belinostat, alongside the vinca alkaloid, vincristine [46]. Vincristine alone, like other spindle toxins, has a propensity to induce some polyploidy alongside mitotic arrest and apoptosis. Belinostat potentiates the apoptotic response. The authors speculate that there are fewer polyploid cells as fewer cells undergo prolonged arrest, mitotic slippage and cytokinesis failure [46]. More cancer cells succumb to acute apoptosis. So, the cooperative effect of these two drugs attacks cells before they have a chance to undergo endoreduplication.
Flavopiridol, a broad spectrum cyclin-dependent kinase (CDK) inhibitor, has also been suggested to reduce the propensity for polyploid cell formation with spindle toxins [47]. This activity is attributed to G1 arrest of cancer cells and occurs even in cells deficient for tumor suppressor genes that abrogate the G1 checkpoint response and which have a propensity for endoreduplication upon treatment with spindle toxins. So, a cytostatic effect of flavopiridol may inhibit endoreduplication and polyploidy elicited by spindle toxins, at least in vitro. The propensity for apoptosis to occur alongside flavopiridol-induced G1 arrest can be dependent on both cell type and drugs that are used alongside this CDK inhibitor (for review see [48]).
Development and maintenance of polyploidy may come with exploitable energy costs. Polyploid cells have increased size and DNA content and sustaining this while initiating new rounds of DNA synthesis requires increased energy input compared to diploid counterparts. As a master regulator of cellular energy use, mechanistic target of rapamycin complex 1 (mTOR1) translates metabolic and environmental cues into a cascade of events that enable anabolic processes such as mRNA translation and lipid synthesis and can limit catabolic processes such as autophagy. The anti-cancer effects of Aurora kinase B inhibitors are enhanced by co-treatment with mTOR inhibitors [49]. Both rapamycin and torkinib (PP242) potentiated Aurora kinase inhibitor-induced apoptosis and induction of autophagic death in polyploid acute myeloid leukemia (AML) cells. Glycolytic metabolism was found to be enhanced in polyploid cells and cooperation was attributed to enhanced metabolic stress [49]. Along similar lines, activation of 5’ AMP-activated protein kinase (AMPK), a direct upstream inhibitor mTOR, by either the natural product, resveratrol, or by salicylate, the active product of Aspirin, can inhibit polyploid cell formation [50]. This occurred alongside treatment with polyploidinducing drugs nocodazole, cytochalasin D or an Aurora kinase B inhibitor. Importantly, the anti-polyploidy activity was validated in vivo using the APCmin model of colorectal cancer [50].
Attacking Existing Polyploid Cancer Cells
Preferentially targeting polyploid cells or preventing the polyploid to aneuploid cell transition could disable tumor progression. High-throughput screening for compounds that selectively kill polyploid cells suggests gene dosage may be an exploitable trait [51]. For example, 8-azaguanine, a compound that requires conversion to a bioactive metabolite by the enzyme hypoxanthine phosphoribosyl transferase 1 (HPRT1), is more toxic to polyploid cancer cells. The extra copies of HRPT1 in polyploid cells underly this toxicity [51]. Altered expression of other genes may also be exploitable. Genes that regulate meiotic cell division have been found to be upregulated in polyploid cancer cells, alongside genes that regulate mitotic division [41,52]. This means cell division, including the reductive divisions that produce aneuploid progeny, either through nuclear budding, multipolar division or other means, may exploit a distinct set of cell division proteins compared to diploids. It is unknown if any meiosis-specific proteins are absolutely required or druggable in polyploid cells, but identifying such vulnerabilities will move a step closer to targeted therapies for polyploid cancer cells.
A distinct strategy has been uncovered using a cell culture system to examine the synthetic lethality between MYC and inhibition of Aurora kinase B. Pro-survival members of the Bcl2 family were confirmed to enable the persistence of polyploid cells [53,54]. Cooperation between Aurora kinase B inhibitors, and inhibitors of pro-survival Bcl2 proteins, has been previously explored [55-57]. Cooperative effects have been assumed due to enhanced apoptosis through activation of the intrinsic apoptotic pathway. However, new findings suggest a distinct mechanism. Pro-survival Bcl2 family proteins also interact with the BH3 only protein Beclin1 (also ATG6) at the endoplasmic reticulum to blockade autophagy [58,59]. This interaction was demonstrated to be crucial for preventing the lethal autophagy that accompanies polyploidy and to contribute to drug resistance in an in vitro model [54]. This research pinpoints a targetable mechanism of action to directly attack polyploid cancer cells. BH3 mimetic drugs disrupt the interaction of pro-survival Bcl2 family proteins with the BH3 domain of Beclin1 and this tactic can be used in combination with drugs such as Aurora kinase inhibitors to enhance cell killing. BH3 mimetics have also been shown to be effective alongside other drugs that induce polyploidy [60].
Other means to disrupt the Beclin1/Bcl2 interaction may also prove valuable. Ceramides are a family of lipids composed of sphingosine and a fatty acid chain. They are found in various cellular membrane compartments, including the Golgi and lysosome and can modify cell signaling pathways. Short-chain ceramides have been found to induce the dissociation of the complex formed between Beclin1 and Bcl2 through the activation of c-Jun N-terminal kinase 1 (JNK1) [61]. JNK1 phosphorylates the Bcl2 protein and this interferes with the association between Beclin1 and Bcl2, thereby enabling autophagy [61]. For polyploid cells, this autophagy is lethal. Knockdown of the gene encoding the ceramide transport protein (known as COL4A3BP or CERT), which moves ceramide from the endoplasmic reticulum (ER) to the Golgi apparatus, induces expression of lysosome-associated membrane protein 2 (LAMP2) and increases autophagic flux, leading to polyploid cell death [62]. So, COL4A3BP may be a target for therapeutic intervention to attack polyploid cancer cells that ultimately works via disruption of the Beclin1/ Bcl2 interaction.
Direct targeting approaches do not only have to target polyploid cells. For example, inhibition of PLK1 alongside treatment with spindle toxins leads to enhanced apoptosis of both diploid and polyploid cancer cells, but polyploid cells have enhanced sensitivity [63]. The enhanced effect of PLK1 inhibition on cells with >4n DNA content was attributed to an inability of polyploid cells to tolerate any further increase in ploidy that was induced by PKL1 inhibition. Polyploid cells were more readily moved toward mitotic catastrophe-induced apoptosis. Genome duplication also increases sensitivity to pharmacological inhibitors of mitotic kinesin family member 11 (also known as Eg5) [64] and monopolar spindle protein 1 (MPS1) [65], so sustained inhibition of mitotic regulators is more toxic to polyploid cells than their diploid counterparts. In theory, these approaches will target both diploid and polyploid cancer cells and could be effective therapies for attacking all cancer cells.
Summary
Genomic instability is a hallmark of cancer and polyploid cells have emerged as an intermediate cell on the path toward aneuploidy. Approaches that prevent the formation of and/or target existing polyploid cancer cells are actively being investigated. However, we are just beginning to understand how to best attack polyploid cancer cells. It appears combination therapies that attack all cancer cells, but due to unique vulnerabilities can preferential impact polyploid cells, may have promise. Enabling lethal autophagy has emerged as one means to attack the polyploid cell population of cancer. Additional research is also required to investigate the role that polyploid cancer cells could play in anti-tumor immunity. The ER stress response and calreticulin play a role in immune surveillance for aberrant polyploidy, but how this is bypassed to enable the persistence of polyploid cancer cells in patients is enigmatic. Therapeutics that reestablish immune attack on polyploid cells, alongside therapeutics that preferentially attack the vulnerabilities of polyploids, may prove a potent combination that halts tumor progression in its tracks.
Acknowledgments
We thank John Huang for critical reading of the text. The illustration was created with BioRender.com.
References
2. Knöfler M, Haider S, Saleh L, Pollheimer J, Gamage TK, James J. Human placenta and trophoblast development: key molecular mechanisms and model systems. Cellular and Molecular Life Sciences. 2019 Sep;76(18):3479-3496.
3. Machlus KR, Italiano JE. The incredible journey: From megakaryocyte development to platelet formation. Journal of Cell Biology. 2013 Jun 10;201(6):785-96.
4. Sagulenko E, Savelyeva L, Ehemann V, Sagulenko V, Hofmann W, Arnold K,et al. Suppression of polyploidy by the BRCA2 protein. Cancer letters. 2007 Nov 8;257(1):65- 72.
5. Aylon Y, Oren M. p53: guardian of ploidy. Molecular oncology. 2011 Aug;5(4):315-23.
6. Dikovskaya D, Schiffmann D, Newton IP, Oakley A, Kroboth K, Sansom O, Jamieson TJ, Meniel V, Clarke A, Nathke IS. Loss of APC induces polyploidy as a result of a combination of defects in mitosis and apoptosis. The Journal of cell biology. 2007 Jan 15;176(2):183-95.
7. Müller A, Gillissen B, Richter A, Richter A, Chumduri C, Daniel PT, et al. Pan-class I PI3-kinase inhibitor BKM120 induces MEK1/2-dependent mitotic catastrophe in non- Hodgkin lymphoma leading to apoptosis or polyploidy determined by Bax/Bak and p53. Cell Death & Disease. 2018 Mar 7;9(3):1-4.
8. Priebe MK, Dewert N, Amschler K, Erpenbeck L, Heinzerling L, Schön MP, et al. c-Rel is a cell cycle modulator in human melanoma cells. Experimental Dermatology. 2019 Feb;28(2):121-8.
9. Zhang S, Chen Q, Liu Q, Li Y, Sun X, Hong L, et al. Hippo signaling suppresses cell ploidy and tumorigenesis through Skp2. Cancer Cell. 2017 May 8;31(5):669-84.
10. Joglekar AP. A cell biological perspective on past, present and future investigations of the spindle assembly checkpoint. Biology. 2016 Dec;5(4):44.
11. Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nature reviews Molecular Cell Biology. 2007 May;8(5):379-93.
12. Rieder CL, Maiato H. Stuck in division or passing through: what happens when cells cannot satisfy the spindle assembly checkpoint. Developmental Cell. 2004 Nov 1;7(5):637-51.
13. Baslan T, Kendall J, Volyanskyy K, McNamara K, Cox H, D’Italia S, Ambrosio F, Riggs M, Rodgers L, Leotta A, Song J. Novel insights into breast cancer copy number genetic heterogeneity revealed by single-cell genome sequencing. Elife. 2020 May 13;9:e51480.
14. Mirzayans R, Andrais B, Murray D. Roles of polyploid/ multinucleated giant cancer cells in metastasis and disease relapse following anticancer treatment. Cancers. 2018 Apr;10(4):118.
15. Coward J, Harding A. Size does matter: why polyploid tumor cells are critical drug targets in the war on cancer. Frontiers in Oncology. 2014 May 26;4:123.
16. Bou-Nader M, Caruso S, Donne R, Celton-Morizur S, Calderaro J, Gentric G, et al. Polyploidy spectrum: a new marker in HCC classification. Gut. 2020 Feb 1;69(2):355- 64.
17. Shimono J, Miyoshi H, Kiyasu J, Kamimura T, Eto T, Miyagishima T, et al. Clinicopathological analysis of polyploid diffuse large B-cell lymphoma. PloS one. 2018 Apr 11;13(4): e0194525.
18. Sidana S, Jevremovic D, Ketterling RP, Tandon N, Greipp PT, Baughn LB, et al. Tetraploidy is associated with poor prognosis at diagnosis in multiple myeloma. American Journal of Hematology. 2019 May;94(5):E117.
19. Borgstrom GH, Vuopio P, de la Chapelle A. Polyploidy of the bone marrow. Scandinavian Journal of Haematology. 1976 Aug;17(2):123-31.
20. Islam S, Paek AL, Hammer M, Rangarajan S, Ruijtenbeek R, Cooke L, et al. Drug-induced aneuploidy and polyploidy is a mechanism of disease relapse in MYC/ BCL2-addicted diffuse large B-cell lymphoma. Oncotarget. 2018 Nov 13;9(89):35875.
21. Bharadwaj D, Mandal M. Senescence in polyploid giant cancer cells: A road that leads to chemoresistance. Cytokine & Growth Factor Reviews. 2020 Apr 1;52:68-75.
22. Zheng L, Dai H, Zhou M, Li X, Liu C, Guo Z, et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nature Communications. 2012 May 8;3(1):1- 2.
23. Kuznetsova AY, Seget K, Moeller GK, de Pagter MS, de Roos JA, Dürrbaum M, et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle. 2015 Sep 2;14(17):2810-20.
24. Mittal K, Donthamsetty S, Kaur R, Yang C, Gupta MV, Reid MD, et al. Multinucleated polyploidy drives resistance to Docetaxel chemotherapy in prostate cancer. British Journal of Cancer. 2017 Apr;116(9):1186-94.
25. Liu K, Lu R, Zhao Q, Du J, Li Y, Zheng M, et al. Association and clinicopathologic significance of p38MAPK-ERK-JNK-CDC25C with polyploid giant cancer cell formation. Medical Oncology. 2020 Jan 1;37(1):6.
26. Rohnalter V, Roth K, Finkernagel F, Adhikary T, Obert J, Dorzweiler K, et al. A multi-stage process including transient polyploidization and EMT precedes the emergence of chemoresistent ovarian carcinoma cells with a dedifferentiated and pro-inflammatory secretory phenotype. Oncotarget. 2015 Nov 24;6(37):40005.
27. Zhang S, Mercado-Uribe I, Xing Z, Sun B, Kuang J, Liu J. Generation of cancer stem-like cells through the formation of polyploid giant cancer cells. Oncogene. 2014 Jan;33(1):116-28.
28. White-Gilbertson S, Voelkel-Johnson C. Giants and monsters: Unexpected characters in the story of cancer recurrence. Advances in Cancer Research. 2020 May 4;148:201-32.
29. Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012 Sep 28;337(6102):1678-84.
30. Gardai SJ, McPhillips KA, Frasch SC, Janssen WJ, Starefeldt A, Murphy-Ullrich JE, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005 Oct 21;123(2):321-34.
31. Krause KH, Michalak M. Calreticulin. Cell. 1997 Feb 21;88(4):439-43.
32. Boilève A, Senovilla L, Vitale I, Lissa D, Martins I, Métivier D, et al. Immunosurveillance against tetraploidization-induced colon tumorigenesis. Cell Cycle. 2013 Feb 1;12(3):473-9.
33. Acebes-Huerta A, Lorenzo-Herrero S, Folgueras AR, Huergo-Zapico L, Lopez-Larrea C, López-Soto A, et al. Drug-induced hyperploidy stimulates an antitumor NK cell response mediated by NKG2D and DNAM-1 receptors. Oncoimmunology. 2016 Feb 1;5(2):e1074378.
34. Castedo M, Coquelle A, Vivet S, Vitale I, Kauffmann A, Dessen P, et al. Apoptosis regulation in tetraploid cancer cells. The EMBO journal. 2006 Jun 7;25(11):2584-95.
35. Castedo M, Coquelle A, Vitale I, Vivet S, Mouhamad S, Viaud S, et al. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. Annals of the New York Academy of Sciences. 2006 Dec;1090(1):35-49.
36. Bojko A, Staniak K, Czarnecka-Herok J, Sunderland P, Dudkowska M, Sliwinska MA, et al. Improved Autophagic Flux in Escapers from Doxorubicin-Induced Senescence/Polyploidy of Breast Cancer Cells. International Journal of Molecular Sciences. 2020 Jan;21(17):6084.
37. Yang D, Liu H, Goga A, Kim S, Yuneva M, Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proceedings of the National Academy of Sciences. 2010 Aug 3;107(31):13836-41.
38. Ogden A, Rida PC, Knudsen BS, Kucuk O, Aneja R. Docetaxel-induced polyploidization may underlie chemoresistance and disease relapse. Cancer Letters. 2015 Oct 28;367(2):89-92.
39. Xuan B, Ghosh D, Cheney EM, Clifton EM, Dawson MR. Dysregulation in actin cytoskeletal organization drives increased stiffness and migratory persistence in polyploidal giant cancer cells. Scientific Reports. 2018 Aug 9;8(1):1-3.
40. Mosieniak G, Sliwinska MA, Alster O, Strzeszewska A, Sunderland P, Piechota M, et al. Polyploidy formation in doxorubicin-treated cancer cells can favor escape from senescence. Neoplasia. 2015 Dec 1;17(12):882-93.
41. Ianzini F, Kosmacek EA, Nelson ES, Napoli E, Erenpreisa J, Kalejs M, et al. Activation of meiosisspecific genes is associated with depolyploidization of human tumor cells following radiation-induced mitotic catastrophe. Cancer Research. 2009 Mar 15;69(6):2296- 304.
42. Sharma S, Yao HP, Zhou YQ, Zhou J, Zhang R, Wang MH. Prevention of BMS-777607-induced polyploidy/ senescence by mTOR inhibitor AZD8055 sensitizes breast cancer cells to cytotoxic chemotherapeutics. Molecular Oncology. 2014 May 1;8(3):469-82.
43. Nair JS, Ho AL, Schwartz GK. The induction of polyploidy or apoptosis by the Aurora A kinase inhibitor MK8745 is p53-dependent. Cell Cycle. 2012 Feb 15;11(4):807-17.
44. Press MF, Xie B, Davenport S, Zhou Y, Guzman R, Nolan GP, et al. Role for polo-like kinase 4 in mediation of cytokinesis. Proceedings of the National Academy of Sciences. 2019 Jun 4;116(23):11309-18.
45. Klauck PJ, Bagby SM, Capasso A, Bradshaw-Pierce EL, Selby HM, Spreafico A, et al. Antitumor activity of the pololike kinase inhibitor, TAK-960, against preclinical models of colorectal cancer. BMC Cancer. 2018 Dec;18(1):1-2.
46. Havas AP, Rodrigues KB, Bhakta A, Demirjian JA, Hahn S, Tran J, et al. Belinostat and vincristine demonstrate mutually synergistic cytotoxicity associated with mitotic arrest and inhibition of polyploidy in a preclinical model of aggressive diffuse large B cell lymphoma. Cancer Biology & Therapy. 2016 Dec 1;17(12):1240-52.
47. Motwani M, Li XK, Schwartz GK. Flavopiridol, a cyclin-dependent kinase inhibitor, prevents spindle inhibitor-induced endoreduplication in human cancer cells. Clinical Cancer Research. 2000 Mar 1;6(3):924-32.
48. Sedlacek HH. Mechanisms of action of flavopiridol. Critical Reviews in Oncology/Hematology. 2001 May 1;38(2):139-70.
49. Liu LL, Long ZJ, Wang LX, Zheng FM, Fang ZG, Yan M, et al. Inhibition of mTOR pathway sensitizes acute myeloid leukemia cells to aurora inhibitors by suppression of glycolytic metabolism. Molecular Cancer Research. 2013 Nov 1;11(11):1326-36.
50. Lissa D, Senovilla L, Rello-Varona S, Vitale I, Michaud M, Pietrocola F, et al. Resveratrol and aspirin eliminate tetraploid cells for anticancer chemoprevention. Proceedings of the National Academy of Sciences. 2014 Feb 25;111(8):3020-5.
51. Choudhary A, Zachek B, Lera RF, Zasadil LM, Lasek A, Denu RA, et al. Identification of selective lead compounds for treatment of high-ploidy breast cancer. Molecular Cancer Therapeutics. 2016 Jan 1;15(1):48-59.
52. Gantchev J, Martínez Villarreal A, Gunn S, Zetka M, Ødum N, Litvinov IV. The ectopic expression of meiCT genes promotes meiomitosis and may facilitate carcinogenesis. Cell Cycle. 2020 Apr 17;19(8):837-54.
53. Zhang J, Zhang S, Shi Q, Yang D, Allen TD. Prosurvival Bcl-2 Proteins are Modifiers of MYC-VX-680 Synthetic Lethality. Journal of Cellular Immunology. 2020 Aug 4;2(5).
54. Zhang J, Zhang S, Shi Q, Allen TD, You F, Yang D. The anti-apoptotic proteins Bcl-2 and Bcl-xL suppress Beclin1/Atg6-mediated lethal autophagy in polyploid cells. Experimental Cell Research. 2020 May 27:112112.
55. Zhang W, Xu J, Ji D, Li Z, He W, Yang F, et al. CyclinG1 amplification enhances aurora kinase inhibitorinduced polyploid resistance and inhibition of Bcl-2 pathway reverses the resistance. Cellular Physiology and Biochemistry. 2017;43(1):94-107.
56. Murai S, Matuszkiewicz J, Okuzono Y, Miya H, De Jong R. Aurora B inhibitor TAK-901 synergizes with BCL-xL inhibition by inducing active BAX in cancer cells. Anticancer Research. 2017 Feb 1;37(2):437-44.
57. Choi JE, Woo SM, Min KJ, Kang SH, Lee SJ, Kwon TK. Combined treatment with ABT-737 and VX-680 induces apoptosis in Bcl-2-and c-FLIP-overexpressing breast carcinoma cells. Oncology Reports. 2015 Mar 1;33(3):1395-401.
58. Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. The EMBO journal. 2007 May 16;26(10):2527-39.
59. Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, et al. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007 Nov 26;3(6):561-8.
60. Zhou W, Xu J, Gelston E, Wu X, Zou Z, Wang B, et al. Inhibition of Bcl-xL overcomes polyploidy resistance and leads to apoptotic cell death in acute myeloid leukemia cells. Oncotarget. 2015 Aug 28;6(25):21557.
61. Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. Journal of Biological Chemistry. 2009 Jan 30;284(5):2719- 28.
62. Lee AJ, Roylance R, Sander J, Gorman P, Endesfelder D, Kschischo M, et al. CERT depletion predicts chemotherapy benefit and mediates cytotoxic and polyploid-specific cancer cell death through autophagy induction. The Journal of Pathology. 2012 Feb;226(3):482-94.
63. Jemaa M, Kifagi C, Serrano SS, Massoumi R. Preferential Killing of Tetraploid Colon Cancer Cells by Targeting the Mitotic Kinase PLK1. Cellular Physiology and Biochemistry. 2020 Apr 1;54(2):303-20.
64. Rello-Varona S, Vitale I, Kepp O, Senovilla L, Jemaá M, Métivier D, et al. Preferential killing of tetraploid tumor cells by targeting the mitotic kinesin Eg5. Cell Cycle. 2009 Apr 1;8(7):1030-5.
65. Jemaà M, Manic G, Lledo G, Lissa D, Reynes C, Morin N, et al. Whole-genome duplication increases tumor cell sensitivity to MPS1 inhibition. Oncotarget. 2016 Jan 5;7(1):885.