Abstract
Sparsely granulated corticotroph pituitary neuroendocrine tumors are rare tumors that may sometimes present without signs of hypercortisolemia, creating diagnostic challenges. We describe a case of a 29-year-old female who sought medical attention due to amenorrhea. Diagnostic evaluations revealed hyperprolactinemia, and an MRI showed a 1.2 x 1.5 x 1.6 cm pituitary macroadenoma, which caused a mass effect on the optic chiasm. The patient subsequently had transsphenoidal endoscopic resection. Histological examination confirmed the presence of a sparsely granulated corticotroph pituitary neuroendocrine tumor with minimal ACTH reactivity. Despite having high serum cortisol and ACTH levels, the patient had minimal clinical signs of Cushing’s disease. This case demonstrates the challenges in diagnosing sparsely granulated corticotroph pituitary adenomas. Without proper identification and management, these tumors can cause significant complications.
Keywords
ACTH-secreting pituitary tumor, Cushing's syndrome, Diagnostic challenge, Sparsely granulated, Corticotroph tumors, Pituitary neuroendocrine tumors
Abbreviations
ACTH: Adrenocorticotropic Hormone; DGCT: Densely Granulated Corticotroph Tumor; FSH: Follicle-Stimulating Hormone; Ki-67: Kiel-67 Proliferation Index; LH: Luteinizing Hormone; POMC: Proopiomelanocortin; SCAs: Silent Corticotroph Adenomas; SGCT: Sparsely Granulated Corticotroph Tumor; TSH: Thyroid-Stimulating Hormone; T-pit: T-box Pituitary Transcription Factor; WHO: World Health Organization.
Introduction
According to the 2022 World Health Organization (WHO) classification, corticotroph adenomas are tumors that originate from adenohypophyseal cells of the T-box pituitary transcription factor (T-PIT) lineage. They produce adrenocorticotropic hormone (ACTH) and other peptides derived from proopiomelanocortin (POMC) [1]. They can be categorized according to their functional activity and histopathological characteristics. Functioning corticotroph adenomas produce excess ACTH, leading to Cushing's disease. In contrast, silent corticotroph adenomas (SCAs) do not cause biochemical or clinical evidence of hypercortisolism, or Cushing's syndrome, such as central obesity, moon face, uncontrolled diabetes, hypertension, and osteoporosis [2].
Histologically, corticotroph adenomas are characterized by positive immunoreactivity for ACTH, which defines them. Based on the granulation pattern within the tumor cells and the distribution of their cytoplasmic keratin filaments, they are subtyped as densely granulated (DGCT), sparsely granulated (SGCTs), and Crooke cell tumors [3]. While DGCTs demonstrate diffuse and intense staining with Periodic Acid-Schiff (PAS) and ACTH, indicating a higher granule density, SGCTs exhibit faint PAS staining and focal, weak ACTH staining, reflecting a lower granule density. The literature indicates that DGCT constitutes the majority of corticotroph tumors; however, SGCTs are larger, more aggressive, and invasive [4,5].
The sparsely granulated subtype may remain undetected for years until it creates symptoms of mass effect. SGCTs, while uncommon, are important to recognize as they can have aggressive features, including apoplexy and greater invasion of surrounding structures, which can lead to their discovery only after significant growth [6]. We report a case of a sparsely granulated corticotroph adenoma that had minimal clinical symptoms of hypercortisolemia.
Case Presentation
A 29-year-old African American woman with a history of well-controlled type 2 diabetes, hypertension, and obesity presented to the emergency department with complaints of lower abdominal pain and concern for possible pregnancy. She reported five months of amenorrhea and two months of intermittent vaginal spotting, as well as left-sided headaches persisting for four months. A pregnancy test returned negative results, leading to a referral to gynecology for further evaluation of amenorrhea. She denied experiencing double or blurry vision, light-headedness, dizziness, focal weakness, or any changes in bladder function. No history of osteoporosis, fragility fractures or proximal muscle weakness was reported.
Investigation
Following her visit to a gynecology clinic, the patient underwent a comprehensive evaluation for amenorrhea (Tables 1 and 2). Work-up revealed elevated prolactin levels, measuring 74 ng/mL (3.34–26.72 ng/mL) in addition to a markedly elevated ACTH level of 105 pg/mL (reference range, 7.2–63.3 pg/mL) One week before surgery, free cortisol was within the normal range (1.46 µg/dL); and total cortisol was not measured preoperatively (Table 2). A subsequent magnetic resonance imaging (MRI) brain revealed a 1.2 x 1.5 x 1.6 cm heterogeneously enhancing pituitary macroadenoma, characterized by a central cystic/necrotic component (Figure 1). The imaging demonstrated mass effect on the optic chiasm and slight extension into the left cavernous sinus, accompanied by a mildly thickened pituitary stalk. As a result, the patient was referred to neurosurgery for surgical intervention.
|
|
16 weeks pre-op |
12 weeks pre-op |
1 week pre-op |
Reference Ranges |
|---|---|---|---|---|
|
PRL |
|
74 ng/mL (non-fasting) |
54 ng/mL (fasting) |
3.34 ng/mL–26.72 ng/mL |
|
ACTH |
|
|
105 pg/mL |
7.2 pg/mL–63.3 pg/mL |
|
LH |
2.94 mIU/mL |
|
|
|
|
FSH |
4.53 mIU/mL |
4.37 mIU/mL |
|
|
|
TSH |
0.71 mIU/mL |
0.62 mIU/mL |
1.78 mIU/mL |
0.45 mIU/mL–5.33 mIU/mL |
|
Free T4 |
0.5 ng/dL |
|
0.47 ng/dL |
0.70 ng/dL–1.70 ng/dL |
|
ACTH: Adrenocorticotropic Hormone; FSH: Follicle-Stimulating Hormone; LH: Luteinizing Hormone; PRL: Prolactin; TSH, Thyroid-Stimulating Hormone; Free T4: Free Thyroxine |
||||
|
|
12 weeks pre-op |
1 week pre-op |
Reference Ranges |
|
Estrogens |
64.3 pg/mL |
|
|
|
Estrone |
46.1 pg/mL |
|
|
|
Testosterone |
47 ng/mL |
|
10 ng/mL–75 ng/mL |
|
Free Testosterone |
0.98 ng/dL |
|
0.04 ng/dL–0.53 ng/dL |
|
Testosterone % Free |
2.1 % |
|
0.4 %- 2.4% |
|
Bioavailable Testosterone |
28.3 ng/dL |
|
1.2 ng/dL–15 ng/dL |
|
Sex Hormone Binding Globulin |
18.5 nMol/L |
|
18.0 nMol/L–135.5 nMol/L |
|
Cortisol -Free |
|
1.46 ug/dL |
|
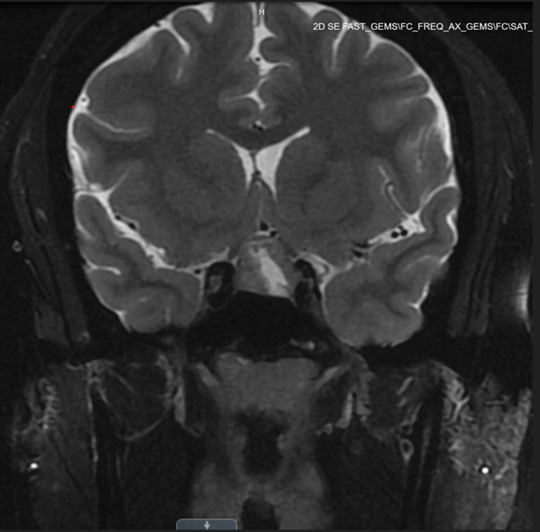
Figure 1. Preoperative pituitary MRI showing pituitary macroadenoma.
Treatment
A transsphenoidal endoscopic resection of the pituitary was performed with a procedural complication of cerebrospinal fluid leak; the otolaryngology team was intraoperatively consulted to repair the leak with no further complications noted.
Outcome and follow-up
Repeat brain MRI on post-operative day 1 showed postsurgical changes of transsphenoidal resection of the pituitary mass with enhancement along the superior and right lateral margin of the resection cavity measuring a maximum of 5 mm, likely representing residual pituitary neoplasm.
Histologic findings were consistent with SGCT, strongly immunoreactive for chromogranin with scattered cells that were lightly immunoreactive for ACTH (Figure 2). No staining was noted for prolactin, growth hormone, follicle-stimulating hormone (FSH), luteinizing hormone (LH), or thyroid-stimulating hormone (TSH). The Ki-67 (Kiel-67) proliferation index labeled about 5–7% of cells in some areas.
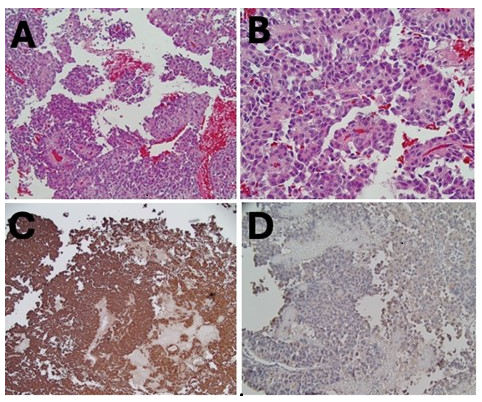
Figure 2. Histological sections of the pituitary tumor. (A, B) Standard hematoxylin-eosin staining. (C) Immunohistochemically stains confirmed strong chromogranin staining. (D) Weak ACTH staining.
Postoperative hormonal evaluation is summarized in Table 3. On postoperative day 1, serum cortisol was markedly elevated. By two weeks after surgery, cortisol had dropped to below the reference range, with ACTH and Prolactin within normal limits.
At two months postoperative follow-up, she reported resolution of headache and resumption of regular menstrual cycles. Anterior pituitary hormonal levels continued to be normal. She is being followed by endocrinology for monitoring of disease recurrence.
|
|
1 day post-op |
2 weeks post-op |
Reference Ranges |
|---|---|---|---|
|
Cortisol |
47.4 ug/dL |
2.4 ug/dL |
6.2–19.4 ug/dL |
|
ACTH |
|
45 pg/mL |
7.2 pg/mL–63/3 pg/mL |
|
Prolactin |
|
20.3 NG/mL |
4.8 NG/mL–33.4 NG/mL |
|
TSH |
|
0.988 uIU/ mL |
0.450 uIU/mL–4.500 uIU/mL |
|
FT4 |
|
0.87 ng/dL |
0.70 ng/dL–1.70 ng/dL |
|
LH |
|
7.5 mIU/mL |
|
|
FSH |
|
5.2 mIU/ml |
|
|
IGF-1 |
|
135 NG/mL |
91 NG/mL - 308 NG/mL |
|
ACTH: Adrenocorticotropic Hormone; FSH: Follicle-Stimulating Hormone; LH: Luteinizing Hormone; PRL: Prolactin; TSH: Thyroid-Stimulating Hormone; Free T4: Free Thyroxine; IGF-1: Insulin-like Growth Factor 1 |
|||
Discussion
This case highlights the challenge related to the clinical classification of corticotroph tumors since our patient’s hormonal data revealed ACTH-dependent hypercortisolism despite the absence of clinical symptoms. The tumor lacked the clinical stigmata of hypercortisolism, with well-controlled hypertension and diabetes; however, elevated ACTH and random cortisol levels suggested some degree of ACTH biological activity. Therefore, this tumor could not be categorized as a silent tumor. SCAs are termed "silent" because they are immunopositive for ACTH but lack biochemical and clinical manifestations of hypercortisolism, such as those seen in Cushing's disease [7,8].
The patient had elevated ACTH and random cortisol levels. Moreover, histopathological examination revealed minimal staining for ACTH. Postoperatively, the ACTH level normalized, and the dexamethasone suppression test demonstrated appropriate suppression. Preoperatively, the presence of obesity, hypertension, and diabetes in our case raised concern for possible hypercortisolemia. However, her blood pressure and diabetes were well controlled with single-agent therapy. While these comorbidities are common in the general population and not exclusive to Cushing disease. Raverot et al., found that the incidence of hypertension and obesity was similar in patients with SCA to that observed in the general population of the same age [9]. Very few similar cases have been reported in the literature of SGCT presenting with biochemical hypercortisolism in the absence of overt Cushingoid characteristics. Although the precise mechanism behind the clinical silence remains unclear, few hypotheses have been postulated to explain the inability of these tumors to develop florid clinical features of hypercortisolemia. A possible explanation is that these tumors produce higher levels of bioinactive ACTH precursors compared with active ACTH, especially since currently available kits cannot differentiate the cleaved bioactive ACTH (1–39) from bioinactive ACTH, pro-ACTH, or POMC [10]. Cases of SCA have been documented with mild elevation of ACTH and normal cortisol and was attributed to high-molecular-weight ACTH with no apparent biological activity [11]. Our patient presented with amenorrhea, which was likely due to hyperprolactinemia attributed to the stalk effect; a phenomenon arising from decreased dopamine inhibition due to suprasellar mass compression, which can lead to mild prolactin elevation. One study found that SGCT mostly presented with symptoms of mass effect, such as headaches, visual field deficits, and hypopituitarism, without clinical features of hypercortisolism [7]. The study included 23 cases of SCAs diagnosed between 1975 and 1997. Among female patients, galactorrhea and amenorrhea were reported in 43% and 29% of cases, respectively. All tumors were large macroadenomas, frequently extending beyond the Sella turcica and exhibiting histopathological features of hemorrhage, necrosis, and cystic degeneration. Despite surgical intervention, more than half of the patients experienced persistent or recurrent disease during long-term follow-up. Similarly, rapidly progressive visual impairment was reported as the most common presentation in another retrospective analysis of 17 cases of surgically resected SCAs [8].
Our patient was diagnosed with a sparsely granulated corticotroph pituitary macroadenoma, a subtype distinguished by certain histological features that significantly impact tumor behavior and prognosis. The SGCTs are often associated with larger tumor size at diagnosis and, in functioning cases, a prolonged duration of Cushing’s disease before diagnosis [3]. SGCTs are associated with aggressive behavior, invasive growth and higher recurrence rates. For instance, Liu et al. reported pituitary apoplexy in a 33-year-old male with a 3.2 cm SGCT, requiring emergent transsphenoidal surgery and resulting in hypopituitarism requiring lifelong hormone replacement [5]. Although our patient lacked overt hypercortisolism or apoplexy, the presence of sparse granulation remains clinically pertinent for its implications on tumor behavior, recurrence risk, and long-term endocrine outcomes. This emphasizes the importance of monitoring plasma ACTH postoperatively, as persistent or rising levels may suggest residual tumor activity or early recurrence. The importance of plasma ACTH levels in the postoperative management of SCAs has been highlighted by Horváth et al., who proposed that monitoring plasma ACTH levels has several advantages [8]. Specifically, it helps in detecting residual tumor tissue even in the absence of clinical symptoms or tumor recurrence via rising ACTH levels. These findings highlight the important role of plasma ACTH monitoring in enhancing long-term management for patients with SCA.
Conclusion
This case emphasizes the importance of a thorough evaluation of pituitary tumors, especially when clinical features are atypical. The distinctive histological features and clinical behavior of SGCTs necessitate tailored management. SGCTs can have invasive growth and therefore, early detection is crucial. A notable limitation in this report was the absence of a routine preoperative workup for hypercortisolemia.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding Statement
This research received no external funding.
Acknowledgments
We would like to thank Dr. Fatiha Grissi from the Department of Pathology for preparing the histopathology images.
Author Contributions Statement
HK wrote most of the background and discussion. JS edited and reviewed the manuscript. SB and SM wrote the case part and prepared the tables. DA helped in writing the discussion and case section.
References
2. Strickland BA, Shahrestani S, Briggs RG, Jackanich A, Tavakol S, Hurth K, et al. Silent corticotroph pituitary adenomas: clinical characteristics, long-term outcomes, and management of disease recurrence. Journal of Neurosurgery. 2021 May 7; 135(6):1706–13.
3. Casar‐Borota O, Burman P, Lopes MB. The 2022 WHO classification of tumors of the pituitary gland: An update on aggressive and metastatic pituitary neuroendocrine tumors. Brain Pathology. 2025 Jan; 35(1):e13302.
4. Doğanşen SÇ, Bilgiç B, Yalin GY, Tanrikulu S, Yarman S. Clinical significance of granulation pattern in corticotroph pituitary adenomas. Turk Patoloji Derg. 2019 Jan 1; 35(1):9–14.
5. Liu T, Rossiter JP, Houlden RL, Awad S. Sparsely granulated corticotroph pituitary macroadenoma presenting with pituitary apoplexy resulting in remission of hypercortisolism. AACE Clinical Case Reports. 2022 Jul 1; 8(4):166–70.
6. Cooper O. Silent corticotroph adenomas. Pituitary. 2015 Apr; 18(2):225–31.
7. Ben-Shlomo A, Cooper O. Silent corticotroph adenomas. Pituitary. 2018 Apr; 21(2):183–93.
8. Fountas A, Lavrentaki A, Subramanian A, Toulis KA, Nirantharakumar K, Karavitaki N. Recurrence of silent corticotroph adenomas after primary treatment: a systematic review and meta-analysis. The Journal of Clinical Endocrinology & Metabolism. 2019 Apr; 104(4):1039–48.
9. Raverot G, Wierinckx A, Jouanneau E, Auger C, Borson-Chazot F, Lachuer J, et al. Clinical, hormonal and molecular characterization of pituitary ACTH adenomas without (silent corticotroph adenomas) and with Cushing's disease. European Journal of Endocrinology. 2010 Jul; 163(1):35–43.
10. Gibson SA, Ray DW, Crosby SR, Dornan TL, Jennings AM, Bevan JS, et al. Impaired processing of proopiomelanocortin in corticotroph macroadenomas. The Journal of Clinical Endocrinology & Metabolism. 1996 Feb 1; 81(2):497–502.
11. Reincke M, Allolio B, Saeger W, Kaulen D, Winkelmann W. A pituitary adenoma secreting high molecular weight adrenocorticotropin without evidence of Cushing’s disease. The Journal of Clinical Endocrinology & Metabolism. 1987 Dec 1; 65(6):1296–300.