Abstract
Background: Guillain-Barré Syndrome (GBS) is a rare but serious autoimmune disorder that typically follows an infection and presents with ascending weakness and areflexia. Systemic Lupus Erythematosus (SLE), a multisystem autoimmune disease, may uncommonly manifest initially with GBS, posing diagnostic and therapeutic challenges.
Case presentation: We report a 25-year-old female who presented with progressive limb weakness diagnosed as acute inflammatory demyelinating polyradiculoneuropathy (AIDP). She failed to respond to standard GBS therapies, including plasmapheresis and IVIG. During hospitalization, she developed features suggestive of systemic involvement including rash, oral ulcers, and nephrotic-range proteinuria. Further investigations revealed positive ANA and anti-dsDNA antibodies, low complement levels, and lupus nephritis (Class V) on renal biopsy, confirming SLE. Immunosuppressive treatment with steroids and mycophenolate mofetil led to clinical improvement.
Conclusion: This case highlights the importance of considering autoimmune etiologies like SLE in patients with atypical or treatment-refractory GBS. Early recognition and initiation of immunosuppressive therapy are essential for optimal neurological and systemic outcomes.
Keywords
Guillain-Barré syndrome (GBS), Systemic lupus erythematosus (SLE), Acute inflammatory demyelinating polyradiculoneuropathy (AIDP), Autoimmune neuropathy
Introduction
Guillain-Barré syndrome (GBS) is an acute, immune-mediated demyelinating neuropathy manifested by rapidly evolving, bilateral limb weakness, areflexia, and varying degrees of sensory and autonomic dysfunction. The most common electrophysiological variant, acute inflammatory demyelinating polyradiculoneuropathy (AIDP), is responsible for approximately 85–90% of GBS cases in North America and Europe [1]. AIDP typically presents with ascending paralysis and is often preceded by an infectious trigger. While GBS is usually monophasic and self-limiting, a typical or recurrent cases may suggest an underlying autoimmune disorder [2,3].
Systemic lupus erythematosus (SLE) is an autoimmune disorder involving every organ system, including the peripheral and central nervous systems. Although SLE is known to affect peripheral and central nerves, GBS as first presentation is uncommon and presents a diagnostic challenge, as described in several case reports and reviews [4,5]. A systematic review by Bhoi et al. in 2023 identified only 27 published cases over the past five decades where GBS preceded or coincided with a diagnosis of SLE, underscoring the diagnostic challenge in such presentation [6].
The pathophysiological link between GBS and SLE remains speculative but is thought to involve autoimmune mechanisms—such as molecular mimicry, autoantibody production, and engagement of the complement pathway—all of which contribute to demyelination and neuronal injury [7–9]. Some researchers have proposed that GBS may represent an initial or early neuro-immunologic manifestation of SLE in predisposed individuals [10]. Recent immunological studies provide further nuance: GBS exemplifies true molecular mimicry at the B-cell level, with infectious epitopes inducing cross-reactive antibodies against neural gangliosides [11]. In SLE, polyclonal B-cell activation, impaired clearance of apoptotic debris, and generation of autoantibodies such as anti-dsDNA and antiphospholipid antibodies create a milieu of immune dysregulation that can also promote neuroinflammation [12]. Complement deposition, particularly of the membrane attack complex (C5b-9), and dysregulated T-cell and cytokine networks (e.g., type I interferons, IL-6) further amplify nerve injury. These overlapping mechanisms provide a plausible explanation for why, in certain predisposed individuals, GBS may not simply co-occur with but herald the onset of SLE.
We describe a case of a young woman presenting with an AIDP variant of GBS, refractory to standard treatment, which was ultimately recognized as a neurological manifestation of underlying SLE. It underscores the importance of considering autoimmune etiologies, such as SLE, in patients presenting with atypical or treatment-resistant GBS [13].
Case Presentation
A 25-year-old female presented with one week history of progressive lower limb weakness that subsequently involved upper limbs. Her past medical history was insignificant and she negated any recent history of diarrhea, respiratory infection or vaccination. There were no bladder and bowel complaints.
Neurological examination revealed flaccid paralysis of all four limbs, flexor plantar responses along with loss of ankle and knee jerks, suggesting a lower motor neuron pathology. However, sensations remained intact.
Keeping in view her examination findings, nerve conduction studies (NCS) were advised which showed AIDP.
An initial diagnosis of GBS was made and she was advised plasmapheresis. Despite receiving 5 sessions of plasmapheresis her weakness didn’t improve. A repeat NCS after 2 weeks was advised which showed severe axonal sensorimotor polyneuropathy with spontaneous activity and no signs of re-innervation suggesting post AIDP sequelae. She was then advised IVIG for 4 days but her clinical condition showed only marginal improvement. While admitted she also developed left leg deep venous thrombosis for which she was commenced on Apixaban.
A detailed clinical examination revealed conjunctival pallor, oral ulcers, non-scarring alopecia, a butterfly shaped malar rash on face, discoid rash on her limbs (Figure 1), generalized pitting edema with left leg being markedly swollen, warm, and tender. Her fundoscopy revealed a single cytoid body at 5’ o clock position in the right eye.
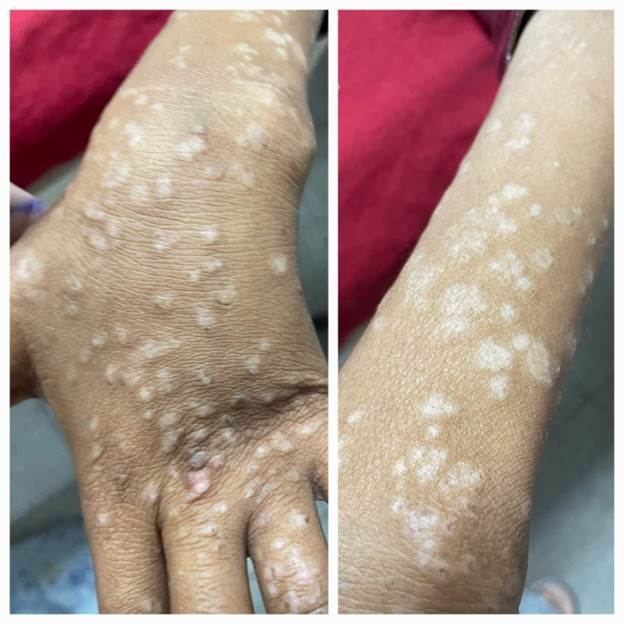
Figure 1. Hypopigmented macules over extensor surfaces, consistent with discoid lupus erythematosus sequelae.
On respiratory examination, breath sounds were diminished and percussion revealed dullness over the right lower lung zone.
Abdomen was distended and tender, with dullness and shifting dullness on percussion.
Investigations showed anemia, thrombocytopenia, a raised ESR of 124 mm/hour, hypoalbuminemia, deranged renal function, Urine routine examination showed microscopic hematuria and proteinuria. Subsequent 24-hour urinary protein quantification confirmed proteinuria of 5,668 mg (Table 1).
|
Complete Blood Count |
Hemoglobin |
7.7 g/dl |
|
Total Leukocyte Count |
5.8 k/uL |
|
|
Platelets |
108 k/uL |
|
|
Reticulocyte count |
0.5% |
|
|
Peripheral smear |
Hemoglobin |
7.3 g/dl |
|
Total Leukocyte Count |
5.9 K/uL |
|
|
Platelets |
123 K/uL |
|
|
Liver Function Tests |
Alanine Transaminase |
7.4 U/L |
|
Alkaline phosphatase |
40 U/L |
|
|
Total Bilirubin |
0.08 mg/dl |
|
|
Coagulation profile |
Prothrombin Time |
14 s |
|
Activated Partial Thromboplastin Time |
36 s |
|
|
International Normalized Ratio |
1 |
|
|
Serum Electrolytes |
Sodium |
135.9 |
|
Potassium |
4.2 |
|
|
Chloride |
110.4 |
|
|
Renal function tests |
Blood urea |
129 mg/dl |
|
Creatinine |
2.07 mg/dl |
|
|
Serum albumin |
0.99 g/dl |
|
|
ESR |
124 mm/hour |
|
|
Urine Routine Examination |
Red Blood Cells |
6–8 |
|
Pus cells |
2–4 |
|
|
|
Protein |
++ |
|
Stool Routine Examination |
Red Blood Cells |
Nil |
|
Pus cells |
Few |
|
|
Cysts |
no cysts |
|
|
Viral profile |
Hepatitis C |
negative |
|
Hepatitis B |
negative |
|
|
Human Immunodeficiency Virus |
negative |
|
|
Lipid profile |
Triglycerides |
132 mg/dl |
|
|
Cholesterol |
201 mg/dl |
|
|
LDL Cholesterol |
133 mg/dl |
|
|
HDL Cholesterol |
43 mg/dl |
|
24-hour Urinary Protein |
|
5,668 mg |
|
Anti-Nuclear Antibodies profile |
|
Positive, 3+ fine speckled Titre :1600 |
|
Nucleosome antibodies |
|
Positive (25AU) |
|
dsDNA antibodies |
|
Positive (25AU) |
|
Histone antibodies |
|
Negative (<0.1AU) |
|
C3 level |
|
13 mg/dl |
|
C4 level |
|
<2.9 mg/dl |
Ultrasound abdomen and pelvis revealed mild abdominopelvic ascites with floating echoes and septations (suggestive of loculated ascites). A right sided pleural effusion was confirmed by chest X-ray.
ECG showed sinus tachycardia; echocardiography revealed mild aortic regurgitation with an ejection fraction of 63% (Table 2). A left lower limb doppler ultrasound also confirmed left leg deep venous thrombosis.
|
Measurement |
Measured |
|
Right Ventricular Internal Diameter (End Diastole) |
20 mm |
|
Septal Thickness |
09 mm |
|
Aortic Root (End Diastole) |
25 mm |
|
Left Ventricular Diastolic Diameter (Diastolic) |
45 mm |
|
Posterior Wall Thickness |
09 mm |
|
Left Ventricular Ejection Fraction |
63% |
|
Left Ventricular Fractional Shortening |
33% |
|
Mild AR documented |
|
A broad differential diagnoses were initially considered. Infectious triggers such as Campylobacter jejuni gastroenteritis were ruled out based on the absence of preceding diarrheal illness. Common respiratory pathogens including Mycoplasma pneumoniae, Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were considered but excluded based on clinical history, lack of systemic symptoms, and negative serologic markers. Post-vaccination GBS was deemed unlikely as there was no recent immunization history. Other differentials such as HIV, paraneoplastic syndromes, and neurotoxic exposures were also excluded based on clinical evaluation, relevant laboratory investigations, and imaging studies.
The patient’s suboptimal response to standard treatment for GBS, significant clinical findings and laboratory evidence of multisystem involvement, prompted further investigation for SLE. Consequently, an autoimmune panel was ordered, which reported the following: ANA Positive, Anti-dsDNA Positive (25AU), Nucleosome antibodies Positive (25 AU), Histone antibodies Negative. Complement levels were decreased; C3: 13mg/dl, C4: 2.5/dl.
A renal biopsy was taken and sent for histopathology, which revealed Lupus Nephritis Class V (Figure 2). Immunofluorescence examination showed variable intensity full house deposits in the glomeruli; IgG deposits (+++) strongly followed by C3(++) and C1q (++) with C1q deposits also present in vessels, tubules and interstation.
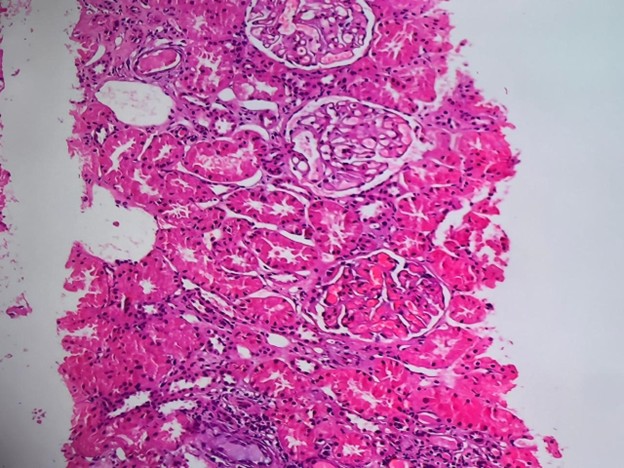
Figure 2. High-power view (H&E stain) highlighting glomeruli with uniform capillary wall thickening and mild mesangial expansion, consistent with membranous features of Class V Lupus Nephritis. No significant endocapillary hypercellularity or necrosis is noted. Activity score: 03, Chronicity score: 02.
A final diagnosis of GBS secondary to SLE was made. Following laboratory and histopathological confirmation of SLE with renal and neurological involvement, the patient was started on immunosuppressive therapy, including intravenous methylprednisolone pulse therapy (1 g daily for 5 days), hydroxychloroquine 200 mg twice daily, mycophenolate mofetil 500 mg twice daily, and ramipril 1.25 mg once daily. Apixaban was continued, and a physiotherapy regimen was initiated.
Follow-up evaluations revealed a marked improvement in her muscle strength as well as in overall disease status.
Discussion
SLE is an autoimmune condition characterized by production of autoantibodies against multiple organs [14]. Mucocutaneous, musculoskeletal, and renal are common manifestations of SLE. Circulating antibodies can also attack the central, peripheral, and autonomic nervous system [15]. Peripheral nervous system is affected in around 10% of all the cases presenting with SLE [16].
GBS is an autoimmune condition-causing limb weakness and is usually due to a bacterial or viral infection [14]. Common etiological causes of GBS include campylobacter jejuni, cytomegalovirus, and Epstein Barr virus (EBV) [17]. The occurrence of GBS as an initial indicator of underlying SLE is rare, with sparse documentation in existing case reports [15]. The prevalence of SLE among GBS patients is reported to be 0.6–1.7%, according to a study by Xiabin et al. [18]. Our case report contributes further to the existing literature.
Most reported cases of GBS as a presenting manifestation of SLE have involved females with a median age of 33 years, which aligns with our case of a 25-year-old female.
The pathophysiological mechanism of GBS in patients with SLE isn't clearly understood and is attributed to cell-mediated and humoral immunity that causes damage to the peripheral nervous system [6]. The patient was initially managed for GBS with intravenous immunoglobulins (IVIG) and plasmapheresis, leading to only mild improvement in muscle weakness. This indicates that IVIG and plasmapheresis alone may not be adequate for managing the SLE-GBS overlap. As demonstrated in our case, additional immunosuppressive therapy is essential for effective treatment [6,15,19]. Our patient significantly improved upon administration of intravenous methylprednisolone pulse therapy, hydroxychloroquine, and mycophenolate mofetil.
The overlap between GBS and SLE, though rare, carries important clinical implications. Previous reports consistently describe patients with limited or absent response to conventional GBS therapies and subsequent improvement only after immunosuppressive treatment, a pattern mirrored in our case. This highlights the need for heightened clinical suspicion when GBS presents atypically or proves refractory to standard therapy. Early recognition of accompanying systemic features such as proteinuria, cytopenias, low complement levels, or cutaneous manifestations should prompt consideration of an underlying autoimmune etiology. Incorporating immunological testing early in the evaluation of such patients may facilitate timely diagnosis and appropriate treatment, ultimately improving neurological and systemic outcomes.
Conclusion
This case emphasizes the importance of considering SLE in the differential diagnosis of GBS, especially in patients with no known triggers or treatment-refractory disease. It also underscores the need for a high index of clinical suspicion and early immunologic screening in cases with atypical neurological presentations, particularly when the response to standard GBS treatment is suboptimal. Moreover, early initiation of treatment for the underlying disease is crucial to improve neurological outcomes and prevent the progression of SLE.
Data Availability
All the relevant data regarding the patient is available upon reasonable request from the corresponding author.
Acknowledgment
None.
Conflict of Interest
The authors have no conflict of interest to declare.
Funding
None.
Patient Consent
Informed consent was signed by the patient’s father.
Authors' Contribution Statement
BJ, MS, ADS, and MHS contributed to the study's conceptualization and methodology, with MO supervising the work. SH, MO, and SHI were responsible for data curation and formal analysis. All authors were involved in writing the original draft and reviewing and editing the manuscript.
References
2. Israeli E, Agmon-Levin N, Blank M, Chapman J, Shoenfeld Y. Guillain–Barré syndrome—a classical autoimmune disease triggered by infection or vaccination. Clinical Reviews in Allergy & Immunology. 2012 Apr;42(2):121–30.
3. Wang Y, Zhao C, Chen R, Ma Z, Tian N, Li X, et al. Case Report: Guillain–Barré syndrome with three episodes and literature review. Frontiers in Immunology. 2025 May 2;16:1559937.
4. Beshir E, Belt E, Chencheri N, Saqib A, Pallavidino M, Terheggen U, et al. Case report: Guillain-Barre syndrome as primary presentation of systemic lupus erythematosus (SLE-GBS) in a teenage girl. Frontiers in Pediatrics. 2022 Mar 17;10:838927.
5. Javadi Parvaneh V, Jari M, Qhasemi S, Nasehi MM, Rahmani K, Shiari R. Guillain–Barre syndrome as the first manifestation of juvenile systemic lupus erythematosus: a case report. Open Access Rheumatology: Research and Reviews. 2019 Apr;24:97–101.
6. Bhoi SK, Jha M, Jaiswal B. Guillain–Barre syndrome as the initial presentation of systemic lupus erythematosus: Case report with a systematic and literature review. Medical Journal Armed Forces India. 2023 Dec 1;79:S360–4.
7. Zhang N, Cao J, Zhao M, Sun L. The introspection on the diagnosis and treatment process of a case of Guillain–Barré syndrome (GBS) attributed to systemic lupus erythematosus (SLE): A case report. Medicine. 2017 Dec 1;96(49):e9037.
8. Hanly JG, Urowitz MB, Su L, Gordon C, Bae SC, Sanchez-Guerrero J, et al. Seizure disorders in systemic lupus erythematosus results from an international, prospective, inception cohort study. Annals of the Rheumatic Diseases. 2012 Sep 1;71(9):1502–9.
9. Bortoluzzi A, Piga M, Silvagni E, Chessa E, Mathieu A, Govoni M. Peripheral nervous system involvement in systemic lupus erythematosus: a retrospective study on prevalence, associated factors and outcome. Lupus. 2019 Apr; 28(4):465–74.
10. Shahrizaila N, Lehmann HC, Kuwabara S. Guillain-barré syndrome. The lancet. 2021 Mar 27;397(10280):1214–28.
11. Laman JD, Huizinga R, Boons GJ, Jacobs BC. Guillain-Barré syndrome: expanding the concept of molecular mimicry. Trends in immunology. 2022 Apr 1;43(4):296–308.
12. Vitale AM, Paladino L, Caruso Bavisotto C, Barone R, Rappa F, Conway de Macario E, et al. Interplay between the chaperone system and gut microbiota dysbiosis in systemic lupus erythematosus pathogenesis: is molecular mimicry the missing link between those two factors?. International Journal of Molecular Sciences. 2024 May 21;25(11):5608.
13. Hughes RA, Cornblath DR. Guillain-barre syndrome. The Lancet. 2005 Nov 5;366(9497):1653–66.
14. Okoh HC, Lubana SS, Langevin S, Sanelli-Russo S, Abrudescu A. A Case of Systemic Lupus Erythematosus Presenting as Guillain‐Barré Syndrome. Case reports in rheumatology. 2015;2015(1):528026.
15. Ali N, Rampure R, Malik F, Jafri SI, Amberker D. Guillain–Barré syndrome occurring synchronously with systemic lupus erythematosus as initial manifestation treated successfully with low-dose cyclophosphamide. Journal of Community Hospital Internal Medicine Perspectives. 2016 Jan 1;6(2):30689.
16. Hanly JG, Urowitz MB, Sanchez‐Guerrero J, Bae SC, Gordon C, Wallace DJ, et al. Neuropsychiatric events at the time of diagnosis of systemic lupus erythematosus: an international inception cohort study. Arthritis & Rheumatism. 2007 Jan;56(1):265–73.
17. Tan H, Caner İ, Deniz O, Büyükavcı M. Miller Fisher syndrome with negative anti-GQ1b immunoglobulin G antibodies. Pediatric neurology. 2003 Oct 1;29(4):349–50.
18. Xianbin W, Mingyu W, Dong X, Huiying L, Yan X, Fengchun Z, et al. Peripheral neuropathies due to systemic lupus erythematosus in China. Medicine. 2015 Mar 1;94(11):e625.
19. Gao Z, Li X, Peng T, Hu Z, Liu J, Zhen J, et al. Systemic lupus erythematosus with Guillian–Barre syndrome: a case report and literature review. Medicine. 2018 Jun 1;97(25):e11160.