Abstract
The electron-dense spherical granules found in the perinuclear region of atrial myocytes store and release both proatrial and probrain natriuretic peptides (proANP and proBNP, respectively). Mature ANP and BNP produce vasodilation and natriuresis and inhibit the renin-angiotensin and sympathetic nervous systems. Although neither ANP nor BNP is a-amidated, Peptidylglycine a-Amidating Monooxygenase (PAM), an integral membrane enzyme known to catalyze the a-amidation of peptidylglycine precursors, is the major atrial granule membrane protein. Selective deletion of PAM from cardiomyocytes impairs their ability to store proANP, resulting in an increase in proANP secretion. Exogenous expression of active or inactive PAM protein restores the ability of atrial myocytes to store proANP, leading to the suggestion that PAM functions as a cargo receptor for newly synthesized proANP.
The Basic Science
The electron-dense spherical granules found in atrial myocytes are one of the most obvious features distinguishing these cells from ventricular myocytes (Figure 1A). In mouse atrial myocytes, granules containing atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) accumulate at one pole of the nucleus, adjacent to a voluminous Golgi complex (Figure 1A) [1,2]. Atrial granules, with their electron dense core and surrounding membrane, look much like the granules that store insulin in islet β-cells and vasopressin in magnocellular hypothalamic neurons. N-terminal signal sequences guide newly synthesized proANP, proinsulin and provasopressin into the lumen of the endoplasmic reticulum (ER), where essential disulfide bonds are formed.
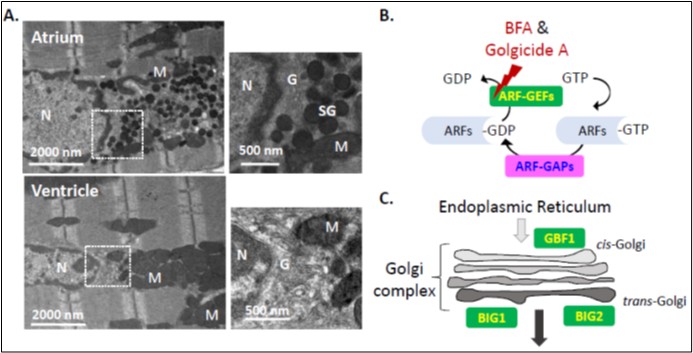
Instead of accumulating near the plasma membrane, ready for release in response to appropriate stimuli, atrial granules remain closely associated with the Golgi complex [2]. The precursors to insulin and vasopressin must undergo a series of prohormone convertase mediated cleavages as they traverse the Golgi complex, exit the trans-Golgi and enter immature secretory granules. Prohormone convertases are not expressed in cardiomyocytes and proANP is stored intact, undergoing Corin-mediated cleavage at the time of secretion [3,4]. Stretch is the major stimulant of ANP release, but endothelin and catecholamines, acting through their G protein coupled receptors, also stimulate ANP release. Calcium entry plays a major stimulatory role in the release of insulin and vasopressin, but has a mild inhibitory effect on ANP secretion [5]. Brefeldin A, a fungal metabolite that disrupts vesicular trafficking by inhibiting Golgi-localized GDP/GTP exchange factors (GEFs) for several ADP ribosylation (ARF) proteins (Figures 1B and 1C), generally inhibits basal secretion. However, pioneering studies on the secretion of proANP by cannulated rat atria and primary cultures of atrial myocytes identified Brefeldin A as a stimulator of basal proANP secretion [6].
Atrial granules contain high levels of proANP and proBNP [1,7]. Surprisingly, the major protein in their membranes is Peptidylglycine a-Amidating Monooxygenase (PAM) [8], a Type 1 integral membrane enzyme that catalyzes the twostep conversion of peptidylglycine substrates into amidated product peptides plus glyoxylate (Figures 2A, and 2B). The copper and ascorbate-dependent monooxygenase domain of PAM (PHM) converts peptidylglycine substrates into peptidyl-a-hydroxyglycine-containing intermediates. The zinc-dependent lyase domain (PAL) converts the penultimate residue of the peptide into an amidated product, releasing glyoxylate (reviewed in [9,10]) (Figures 2A). While many peptides (e.g. vasopressin, gastrin, neuropeptide Y, thyrotropin releasing hormone) are inactive until amidated by PAM, neither proANP, ANP, proBNP, nor BNP is amidated. Early studies revealed high levels of PAM in the atrium, with transient expression of PAM in the ventricles during development [11], but its function in cardiac tissue has remained a mystery for 40 years.
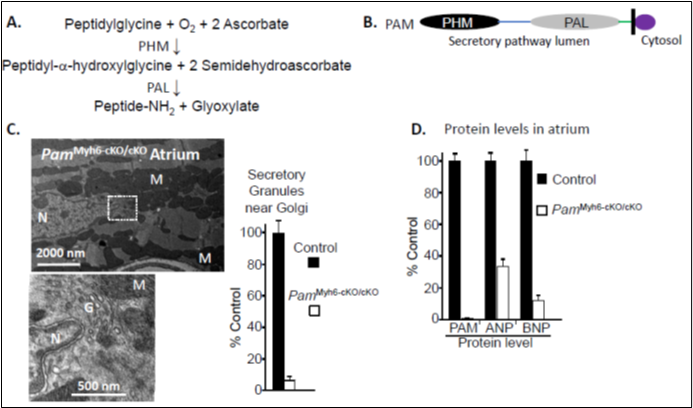
Genetic manipulations that eliminate PAM expression in mice, flies, and zebrafish are not compatible with life [10]. To explore the role of PAM in the heart, mice with a floxed allele of PAM (PamcKO/cKO) were mated to mice in which the expression of Cre-recombinase was driven by the cardiomyocyte-specific myosin heavy chain 6 (Myh6) promoter (PamMyh6/+;cKO/cKO). Mice unable to express PAM in their cardiomyocytes (PamMyh6-cKO/cKO) are viable [12]. Electron microscopy revealed the almost total absence of secretory granules in the atria of PamMyh6-cKO/cKO mice (Figure 2C), with few other changes in their ultrastructure [13]. Atrial levels of proANP and proBNP protein declined in the absence of PAM (Figure 2D).
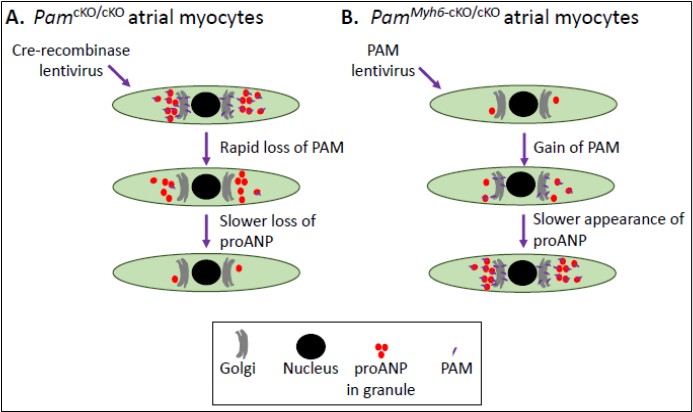
Using primary cultures of atrial myocytes from PamcKO/cKO mice and lentiviral delivery of Cre-recombinase, we established that loss of PAM caused a reduction in proANP content (Figure 3A) [13]. Using primary cultures of atrial myocytes from PamMyh6-cKO/cKO mice and lentiviral delivery of PAM, we demonstrated that expression of PAM in cells which had not previously expressed PAM was sufficient to rescue proANP content to near normal (Figure 3B). PAM lacking one of its essential copper binding sites is enzymatically inactive; strikingly, expression of inactive PAM also restored the proANP content of PamMyh6-cKO/cKO atrial myocytes to near normal (Figure 3B) [13].
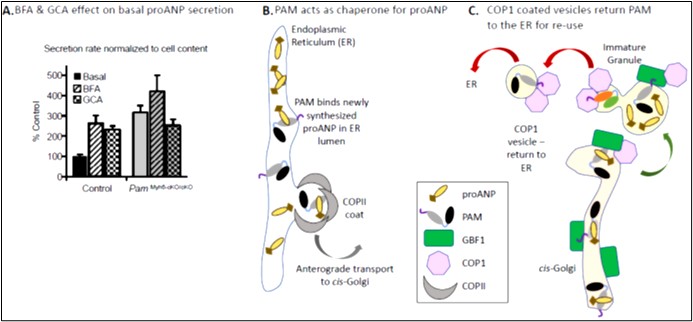
Neither proANP mRNA levels nor proANP bio9s ynthesis differed in wildtype and PamMyh6-cKO/cKO atrial myocytes. However, a 3-fold increase in the basal secretion of proANP was observed in PamMyh6-cKO/cKO atrial myocytes, identifying increased secretion as a major cause of the observed decrease in proANP storage (Figure 4A). Consistent with earlier studies, BFA produced a 3-fold increase in the basal secretion of proANP by control atrial myocytes (i.e. a failure of storage); in contrast, BFA had no significant effect on proANP secretion by atrial myocytes lacking PAM. Golgicide A, which inhibits the GDP/GTP exchange activity of the only cis-Golgi localized ARF-GEF (GBF1) without affecting the trans-Golgi localized ARF-GEFs (Figure 1C), mimicked the effects of BFA (Figure 4A). GBF1 plays an essential role in the formation and trafficking of the COPIcoated vesicles that return cargo from the cis-Golgi to the ER. Taken together, our data support a model in which the PAM protein interacts directly with newly synthesized proANP (Figure 4B) [14], facilitating its transfer from the ER to the cis-Golgi and nascent granules (Figure 4C). With an ~30-fold molar excess of proANP over PAM, GBF1-dependent recycling of PAM plays an essential role in sustaining the efficient delivery of proANP to nascent granules [8]. The activity of its monooxygenase domain is not essential for PAM to play this role. As for several other secretory pathway cargo proteins, the successful storage of large amounts of proANP in granules requires the recycling of its chaperone [15]. A detailed discussion of PAM trafficking in the secretory and endocytic pathways and a comparison of atrial granules to lysosome-related organelles can be found in the review by Bäck et al. [9].
The Clinical Implications
The lack of atrial secretory granules and reduced serum levels of ANP seen in PamMyh6-cKO/cKO mice raise several questions about the consequences of PAM deficiency for the paracrine and endocrine roles of ANP. First and foremost, complete lack of ANP or its principal receptor (NPR1, also designated NPR-A) in mice leads to salt-dependent hypertension and hypertrophy with concomitant fibrosis in the heart [16,17]. The apparent lack of a similar cardiovascular phenotype in the PamMyh6- cKO/cKO mice [12] suggests that the increased basal secretion of proANP, which is thought to occur via a Golgi bypass pathway in isolated PamMyh6-cKO/cKOatrial cardiac myocytes, is sufficient to counteract these changes. Neither plasma levels of BNP nor secretion of BNP by PamMyh6-cKO/cKO atrial myocytes in culture has been assessed, but atrial proBNP was even more depleted than proANP [12,13]. BNP is expressed in both atrial and ventricular myocytes in mice (in a ratio of approx. 1:1) [18]; secretory granules are rarely seen in adult ventricular myocytes, consistent with the fact that ventricular secretion of BNP is constitutive (not regulated). The consequences of the absence of atrial granules in PamMyh6-cKO/cKO mice may first manifest itself during stress-induced, pulsatile release of natriuretic peptides in response to disease, e.g. increased atrial stretch or ischemia, as previously described [19,20].
In animal models of ischemia-reperfusion injury, infusion of ANP or BNP has been shown to redu1c0e the area at risk and infarct size [21] and both ANP and BNP are used clinically to treat cardiac disease [22,23]. Natriuretic peptides have been pursued as potential therapeutics for acute heart failure, although with mixed results due to side effects [24,25]. The clinical use of peptides as drugs may work best without the severe hemodynamic lability seen in heart failure. For example, resistant/essential hypertension may be an ideal diagnosis to begin a trial of natriuretic peptide treatment [26,27]; the cardiospecific PAM knockout models could be an ideal animal model to evaluate the efficacy of peptide treatment, as the endogenous hormonal axis is not capable of functioning normally. Notably, it is well known that mutations in Corin, the key processing enzyme that generates bioactive ANP and BNP from their precursors, are strongly associated with hypertension [28,29]. Whether the absence of PAM affects the ability of Corin to cleave pronatriuretic peptides released from the classical or Golgi bypass pathways has not yet been determined. Finally, whether the lack of pronatriuretic-peptide-containing granules will increase infarct size seems worthy of further mechanistic studies.
The production and release of natriuretic peptides is markedly upregulated during hormonal, hemodynamic, or oxygen-related cardiac stress [22]. However, it is not yet known how PAM deficiency affects the compensatory upregulation seen in hypertension, heart failure, and atrial fibrillation. PAM deficiency has been implicated in a number of human ailments, notably type II diabetes and hypertension [30-33]. Additional important aspects to address include the effects of lack of PAM on Corin trafficking and on BNP processing and secretion. Physiological and mechanistic studies in the cardio-specific PAM knock-out mouse model should prove to be of great value for better understanding the interplay between ANP and BNP in disease and elucidating how changes in the pulsatile release of natriuretic peptides affects the heart and circulation under stress.
Acknowledgments
We thank Maya Yankova for ultrastructural images of mouse heart. This work was supported by NIH Grant DK032948 (to R.E.M.), the Rodney and Janice Reynolds Endowment (B.A.E.), the Daniel Schwartzberg Fund (R.E.M. and B.A.E.).
References
2. Jamieson JD, Palade GE. Specific granules in atrial muscle cells. The Journal of Cell Biology. 1964 Oct 1;23(1):151-72.
3. Yan W, Wu F, Morser J, Wu Q. Corin, a transmembrane cardiac serine protease, acts as a pro-atrial natriuretic peptide-converting enzyme. Proceedings of the National Academy of Sciences. 2000 Jul 18;97(15):8525-9.
4. Glembotski CC, Irons CE, Sprenkle AB, Sei CA. Studies of ANF processing and secretion using a primary cardiocyte culture model. Canadian Journal of Physiology and Pharmacology. 1991 Oct 1;69(10):1525-36.
5. Uusimaa PA, Hassinen IE, Vuolteenaho OL, Ruskoaho HE. Endothelin-induced atrial natriuretic peptide release from cultured neonatal cardiac myocytes: the role of extracellular calcium and protein kinase-C. Endocrinology. 1992 May 1;130(5):2455-64.
6. Ogawa T, Vatta M, Bruneau BG, de Bold AJ. Characterization of natriuretic peptide production by adult heart atria. American Journal of Physiology-Heart and Circulatory Physiology. 1999 Jun 1;276(6):H1977-86.
7. Takemura G, Takatsu Y, Doyama K, Itoh H, Saito Y, Koshiji M, et al. Expression of atrial and brain natriuretic peptides and their genes in hearts of patients with cardiac amyloidosis. Journal of the American College of Cardiology. 1998 Mar 15;31(4):754-65.
8. O’Donnell PJ, Driscoll WJ, Bäck N, Muth E, Mueller GP. Peptidylglycine-a-amidating monooxygenase and proatrial natriuretic peptide constitute the major membraneassociated proteins of rat atrial secretory granules. Journal of Molecular and Cellular Cardiology. 2003 Aug 1;35(8):915-22.
9. Bäck N, Mains RE, Eipper BA. Organism and Cell Type Specific Uses of Peptidylglycine a-Amidating Monooxygenase (PAM). The FEBS Journal, 2021, Jun5.
10. Kumar D, Mains RE, Eipper BA. 60 YEARS OF POMC: From POMC and a-MSH to PAM, molecular oxygen, copper, and vitamin C. Journal of Molecular Endocrinology. 2016 May 1;56(4):T63-76.
11. Quafik LH, May V, Keutmann HT, Eipper BA. Developmental regulation of peptidylglycine a-amidating monooxygenase (PAM) in rat heart atrium and ventricle. Journal of Biological Chemistry. 1989;264:5839-45.
12. Powers KG, Ma XM, Eipper BA, Mains RE. Identifying roles for peptidergic signaling in mice. Proceedings of the National Academy of Sciences. 2019 Oct 1;116(40):20169- 79.
13. Bäck N, Luxmi R, Powers KG, Mains RE, Eipper BA. Peptidylglycine a-amidating monooxygenase is required for atrial secretory granule formation. Proceedings of the National Academy of Sciences. 2020 Jul 28;117(30):17820- 31.
14. Labrador V, Brun C, Ko¨nig S, Roatti A, Baertschi AJ. Peptidyl-glycine a-amidating monooxygenase targeting and shaping of atrial secretory vesicles: inhibition by mutated N-terminal ProANP and PBA. Circulation Research. 2004 Dec 10;95(12):e98-109.
15. Béthune J, Wieland FT. Assembly of COPI and COPII vesicular coat proteins on membranes. Annual Review of Biophysics. 2018 May 20;47:63-83.
16. John SW, Krege JH, Oliver PM, Hodgin JB, Pang SC, Flynn TG, et al. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Science. 1995 Feb 3;267(5198):679-81.
17. Lopez MJ, Wong SK, Kishimoto I, Dubois S, Mach V, Friesen J, et al. Salt-resistant hypertension in mice lacking the guanylyl cyclase-A receptor for atrial natriuretic peptide. Nature. 1995 Nov;378(6552):65-8.
18. Goetze JP, Georg B, Jørgensen HL, Fahrenkrug J. Chamber-dependent circadian expression of cardiac natriuretic peptides. Regulatory Peptides. 2010 Feb 25;160(1-3):140-5.
19. Skvorak JP, Sutton ET, Rao PS, Dietz JR. Mechanism of anoxia-induced atrial natriuretic peptide release in the isolated rat atria. American Journal of Physiology- Regulatory, Integrative and Comparative Physiology. 1996 Jul 1;271(1):R237-43.
20. Dietz JR. Release of natriuretic factor from rat heartlung preparation by atrial distension. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology. 1984 Dec 1;247(6):R1093-6.
21. Burley DS, Baxter GF. B-type natriuretic peptide at early reperfusion limits infarct size in the rat isolated heart. Basic Research in Cardiology. 2007 Nov 1;102(6):529.
22. Goetze JP, Bruneau BG, Ramos HR, Ogawa T, de Bold MK, Adolfo J. Cardiac natriuretic peptides. Nature Reviews Cardiology. 2020 Nov;17(11):698-717.
23. Sridharan S, Kini RM, Richards AM. Venom natriuretic peptides guide the design of heart failure therapeutics. Pharmacological Research. 2020 May 1;155:104687.
24. Ogiso M, Isogai T, Okabe Y, Ito K, Tsuji M, Tanaka H. Effect of carperitide on in-hospital mortality of patients admitted for heart failure: propensity score analyses. Heart and Vessels. 2017 Aug;32(8):916-25.
25. Hata N, Seino Y, Tsutamoto T, Hiramitsu S, Kaneko N, Yoshikawa T, et al. Effects of carperitide on the long-term prognosis of patients with acute decompensated chronic heart failure the PROTECT multicenter randomized controlled study. Circulation Journal. 2008;72(11):1787- 93.
26. Chen Y, Schaefer JJ, Iyer SR, Harders GE, Pan S, Sangaralingham SJ, et al. Long-term blood pressure lowering and cGMP-activating actions of the novel ANP analog MANP. American Journal of Physiology- Regulatory, Integrative and Comparative Physiology. 2020 Apr 1;318(4):R669-76.
27. Buglioni A, Burnett Jr JC. New pharmacological strategies to increase cGMP. Annual Review of Medicine. 2016 Jan 14;67:229-43.
28. Li H, Zhang Y, Wu Q. Role of corin in the regulation of blood pressure. Current Opinion in Nephrology and Hypertension. 2017 Mar;26(2):67-73.
29. Armaly Z, Assady S, Abassi Z. Corin: a new player in the regulation of salt–water balance and blood pressure. Current Opinion in Nephrology and Hypertension. 2013 Nov 1;22(6):713-22.
30. Huyghe JR, Jackson AU, Fogarty MP, Buchkovich ML, Stancáková A, Stringham HM, et al. Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nature Genetics. 2013 Feb;45(2):197-201.
31. Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, et al. Identification of lowfrequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nature Genetics. 2014 Mar;46(3):294-8.
32. Thomsen SK, Raimondo A, Hastoy B, Sengupta S, Dai XQ, Bautista A, et al. Type 2 diabetes risk alleles in PAM impact insulin release from human pancreatic ß-cells. Nature Genetics. 2018 Aug;50(8):1122-31.
33. Yoo HJ, Kim M, Kim M, Chae JS, Lee SH, Lee JH. The peptidylglycine-a-amidating monooxygenase (PAM) gene rs13175330 A> G polymorphism is associated with hypertension in a Korean population. Human Genomics. 2017 Dec;11(1):1-8.