Abstract
Bromodomain-containing protein 4 (BRD4) belongs to the bromodomain and extra-terminal (BET) family of proteins, which regulate gene expression by binding to acetylated histones and transcription factors and then recruiting other transcriptional regulators. The discovery and development of BET inhibitors have greatly advanced our understanding of the cellular effects of BET proteins and their therapeutic relevance to different diseases. In the field of cardiovascular research, BET bromodomain inhibition by small molecules has been investigated and has demonstrated promising effects on various heart diseases. Whereas results from these preclinical investigations have shown promise, clinical trials are still in early phases at this time. In this review, considering the rapidly expanding evidence for targeting BET family proteins in heart disease, we will summarize the preclinical and clinical investigations of the therapeutic potential of BET bromodomain inhibition in heart disease, with a focus on heart failure and diabetic cardiomyopathy.
Keywords
BET inhibition, BRD4, Diabetic cardiomyopathy, Heart failure, Mitochondrial homeostasis
Introduction
Bromodomain-containing protein 4 (BRD4) is a member of the mammalian bromo- and extra-terminal domain (BET) protein family, which also comprises BRD2, BRD3, and testis-specific BRDt. The BET family of proteins shares the structure of two evolutionarily conserved tandem N-terminal bromodomains, as well as an extra-terminal (ET) domain (Figure 1). They bind acetylated histone tails through bromodomains, which results in localization to the chromosome, where they recruit other regulatory complexes, such as transcription elongation factor b (P-TEFb) and mediators, to influence gene expression [1,2]. In 2010, two groups independently discovered JQ1 and I-BET as BET bromodomain inhibitors, a class of small molecules that forms monovalent interactions with individual BET bromodomains that compete for binding of bromodomains to their natural ligand, acetylated lysine [3,4]. Since then, BET bromodomains have become therapeutic targets for designing small anticancer molecules. In cardiovascular research, accumulating evidence shows that pharmacological inhibition of BET proteins has therapeutic effects on heart disease, hypertension [5], pulmonary arterial hypertension [6], and atherosclerosis [7]. In this mini-review, we will specifically summarize the recent advances in BET inhibition in the field of heart disease, with a focus on heart failure (HF) and diabetic cardiomyopathy (DC). We will also discuss the relationship between BRD4 and mitochondrial homeostasis in the heart, the difference between pharmacological BET inhibition and genetic Brd4 knockout mouse models, and the translational potential of BET inhibition for treating human heart disease.
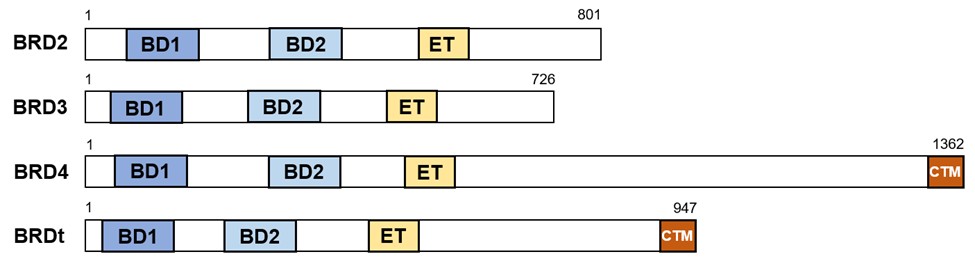
Figure 1. Scheme showing domain architecture of human BET family proteins. The bromodomain and extra-terminal domain (BET) family of proteins comprises BRD2, BRD3, BRD4, and BRDt. Only BRD4 and BRDt have a C-terminal motif. BD: Bromodomain; ET: Extra-Terminal domain; CTM: C-Terminal Motif.
BRD4 Inhibition Attenuates HF
HF can be characterized as a progressive disorder that is initiated after “damage signals”, such as myocardial infarction (MI) and vascular disease–induced interruption of cardiac blood supply, which results in reduced contractile function and inability of the heart to pump sufficient blood to meet bodily demands. HF remains a major cause of morbidity and mortality worldwide [8]. To compensate for adverse hemodynamics and to increase cardiac output during HF, the heart undergoes cardiac hypertrophy, which may be adaptive initially but becomes maladaptive if sustained [9]. Two groups generated cardiac hypertrophy mouse models by transverse aortic constriction (TAC) and found that BET bromodomains inhibition by JQ1 blocked both agonist-dependent hypertrophy in cultured neonatal rat ventricular myocytes and rescued TAC-induced pathologic cardiac remodeling and cardiac dysfunction. Mechanistically, they found BET proteins could be enriched at super-enhancers and acted as essential transcriptional coactivators of pathologic genes that link to cardiomyocyte hypertrophy and HF [10,11]. For the first time, they depicted the role of BET bromodomain proteins in the heart. This dynamic enrichment of BRD4 at superenhancers was regulated by microRNA-9 [12]. Moreover, Duan et al. extended the therapeutic relevance of the work by treating mice with pre-established HF or massive MI with JQ1 and explored the mechanism using humaninduced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). Their integrated transcriptomic analyses across animal models and human iPSC-CMs indicated that BET inhibition blocks transactivation of genes involved in pro-fibrosis and innate inflammatory responses [13].
The heart is a complex organ comprising different cell types. Because JQ1 is administered systemically, it may directly affect not only cardiomyocytes but also noncardiomyocyte populations. For example, Stratton and Bagchi et al. identified BRD4 as a crucial mediator of fibroblast activation in the heart by redistributing to enhancers and super-enhancers in cardiac fibroblasts and promoting the expression of fibrotic genes upon stimulation. BRD4 inhibition by JQ1 blocked this process [14]. Despite protecting against cardiac fibrosis, BET inhibitors have exhibited profound anti-fibrotic effects in various rodent models of organ failure, which has been reviewed in-depth elsewhere [15]. One recent study revealed the anti-inflammatory effect of BET inhibition in a cytokine storm (CS)-induced cardiac dysfunction animal model and multi-cellular human pluripotent stem cell-derived cardiac organoids (hCOs). They highlighted fibroblasts as crucial mediators in inflammationinduced cardiac dysfunction, as the CS induced a more pronounced transcriptional response in fibroblasts than in cardiomyocytes within hCOs [16]. Collectively, these studies establish BET bromodomains inhibition as a viable therapeutic strategy to attenuate acquired forms of HF.
The damage signal that triggers HF is not only acquired, such as MI and TAC, but may be hereditary, as in the case of genetic cardiomyopathies. Dilated cardiomyopathy (DCM) increases the risk of heart failure and sudden cardiac death [17]. Recent studies highlighted the therapeutic effect of BET bromodomains inhibition on mouse models of heritable DCM. Mutations in the LMNA gene, which encodes lamin A/C, have been linked to a variety of diseases, including DCM [18]. Using Lmna cardiomyocyte-specific knockout mice (Lmna-cKO), Auguste et al. discovered increasing Brd4 transcript levels without changes in protein expression. Treating 2-week-old mice with JQ1 for 7 days, the authors observed potent improvements in survival, pathologic cardiac remodeling, and cardiac function. RNA-Seq of primary cardiomyocytes isolated from JQ1-treated mice revealed that JQ1 partially restored the dysregulated transcriptome and secretome of Lmna deficient cardiomyocytes to the level of healthy controls [19]. A dominant Arg-to-Cys (Arginine-to-Cysteine) missense mutation in phospholamban (PLNR9C) is another known genetic cause of human DCM [20]. A study from the Burke group also highlighted the important role of BRD4 in the pathogenesis of DCM using a PLNR9C mouse model. In that work, BET inhibition by JQ1 abrogated adverse cardiac remodeling, reduced cardiac fibrosis, and prolonged survival in PLNR9C mice. Importantly, the authors revealed that BRD4 serves as a direct regulator of NF-κB–mediated proinflammatory gene expression in cardiac fibroblasts but not in adult cardiomyocytes [21]. Taken together, these studies implicate the activation of BET proteins in the pathogenesis of heritable HF and uncovered a potential therapeutic strategy for this condition by BET bromodomains inhibition.
BRD4 Inhibition Protects Against DC
DC develops in the absence of traditional risk factors, such as hypertension and coronary artery disease in patients with diabetes. According to the United States Centers for Disease Control and Prevention (CDC), type 2 diabetes mellitus (T2DM) is the most common form of diabetes, affecting 90% to 95% of total patients. The risk of HF in men and women with T2DM is at least 2.4-fold and 5.1- fold higher, respectively, than in sex-matched individuals without diabetes [22]. Despite the fact that cardiac complications are elevated in patients with diabetes, effective therapeutic strategies for diabetes-associated cardiovascular disease prevention and treatment remain elusive. Streptozotocin (STZ) is particularly toxic to the insulin-producing beta cells of the pancreas in mammals and is commonly used to induce rodent models of type 1 diabetes. Given that JQ1 improves cardiac function and pathologic cardiac remodeling in mouse models of TAC and MI, Guo et al. revealed that JQ1 also reduced STZ-induced cardiac fibrosis and cardiomyocyte apoptosis through caveolin-1, a negative regulator of TGF-β1 (transforming growth factor-β1) signaling [23].
Because T2DM has strong association with the prevalence of cardiac complications, investigating BET bromodomains inhibition in T2DM animal models is important for understanding this relationship. A recent study from our group discovered aberrant upregulation of BRD4 protein expression in high-fat-diet (HFD)-induced DC. Inhibition of BRD4 by chronic JQ1 administration alleviated DC disease progression, as evidenced by complete restoration of normal cardiac diastolic and systolic function and nearcomplete prevention of cardiac hypertrophy, interstitial fibrosis, lipid accumulation, and cardiomyocyte apoptosis induced by 6-month HFD-feeding [24]. However, in this study, we only focused on the effect of JQ1 in late-stage DC, which is characterized by systolic cardiac dysfunction with adverse structural remodeling. Exploring whether JQ1 has a preventive effect on pathogenesis of earlystage DC, which is characterized by diastolic cardiac dysfunction, is of great interest. Moreover, T2DM is often associated with insulin resistance (IR), which occurs in many tissues, including skeletal muscle, liver, and heart. The common insulin-sensitizing therapy, Rosiglitazone, however, significantly increases the risk of cardiac damage [25]. We found that JQ1 ameliorated diabetes-induced IR, as evidenced by improved glucose and insulin tolerance (unpublished data). Taken together, our findings indicate that BET bromodomains inhibition is not only a potential therapeutic approach for DC but could also be used as an insulin sensitizer without cardiac side effects to alleviate metabolic syndrome.
BRD4 Regulates Mitochondrial Homeostasis in the Heart
The heart is the most metabolically active organ in the body. To meet the enormous energy requirement for contraction and relaxation, the heart possesses the highest content of mitochondria in the body, which compose 25%–30% of the cell volume across mammalian species, compared to any other organs [26]. Given that considerable evidence of altered mitochondrial energetics has been observed in HF [27] and that BET bromodomains inhibition demonstrably improves heart function in several HF and DC mouse models, specific studies that target the regulatory effect of BRD4 on mitochondrial homeostasis will be of great interest. Dysfunctional mitochondria can be sequestered and delivered to lysosomes for degradation during mitophagy. PTEN-induced putative kinase protein 1 (PINK1) is a serine/threonine kinase that is localized at the mitochondrial outer membrane and mediates mitophagy in a ubiquitination-dependent manner [28]. A recent study from our group revealed that JQ1 improved dysfunctional mitochondrial clearance by activating PINK1-mediated mitophagy in the diabetic heart. We found enrichment of both BRD4 and acetylated histone 3 lysine 27 (H3K27ac) at the Pink1 promoter in hearts of HFD-fed mice, implying that BRD4 binds to H3K27ac at the Pink1 promoter region. Deletion of Pink1 abrogates the beneficial effect of JQ1 on cardiac diastolic and systolic functions, cardiac lipid accumulation, and cardiomyocyte apoptosis [24].
A recent elegant study by Padmanabhan et al. revealed a different phenotype of a Brd4 genetic deletion mouse model. Unlike pharmacological inhibition, cardiomyocytespecific deletion of Brd4 in adult mice leads to acute deterioration of cardiac contractile function. To delineate the underlying mechanism, they found that BRD4 and GATA4, a well-established lineage-determining transcription factor of cardiomyocytes, co-localized and formed a novel protein complex at gene loci relevant to mitochondrial bioenergy production. Importantly, this complex is bromodomain-independent, as BRD4 bromodomains acetylated-lysine interaction-defective mutant constructs retained the ability to interact with GATA4 [29]. Their study first elucidated the critical role of BRD4 in regulating cardiomyocyte mitochondrial homeostasis. In addition, they highlighted the structurefunction relationships of BRD4 outside of bromodomains. Kim et al. similarly found that a genetic knockout of Brd4 triggered progressive decline in myocardial function, culminating in DCM due to significant loss of mitochondrial electron transport chain protein expression and activity [30]. Taken together, these studies of genetic knockout mouse models elucidated a novel role for BRD4 in regulating cardiomyocyte mitochondrial homeostasis, which is indispensable for normal heart function.
Pharmacological BET Inhibition vs. Genetic Brd4 Knockout
Notably, the Brd4 cardiomyocyte-specific knockout mouse model displayed an outcome distinct from pharmacological BET inhibition. Here, we summarize our thoughts on the possible causes of these disparities. First, BET bromodomains inhibitors engage at the bromodomain pocket of BET proteins competitively with acetylated-lysine binding, thereby displacing BET proteins from chromatin. Other BET family proteins, such as BRD2 and BRD3, can also be influenced by pharmacological BET inhibition. Second, in addition to acetylated-lysine binding bromodomains, BRD4 comprises several other domains that are similarly important for transcription factor interaction and chromatin regulation (Figure 2). For example, the ET domain of BRD4 can regulate gene transcription by interacting with several histone modifiers, such as the arginine demethylase JMJD6 and the lysine methyltransferase NSD3 [31,32]. The BID and PDID domains of BRD4 interact directly with p53 [33]. In addition, BRD4 has been characterized as a histone acetyltransferase that acetylates H3K122, which plays a critical role in nucleosome stability [34]. One recent study indicated that prolyl hydroxylase domain protein 2 (PHD2)- mediated proline hydroxylation of BRD4 significantly affects its interaction with key transcription factors, as well as BRD4-mediated transcriptional activation [35,36]. Unlike genetic Brd4 knockout, pharmacological BET inhibition only partially affected BRD4 function. Finally, BET inhibition is systemic, so the beneficial effect of BET inhibition on heart disease may be due to a combination effect on multiple cell types. Overall, genetic modulation is an entirely different molecular perturbation than smallmolecule inhibition of BET bromodomains.
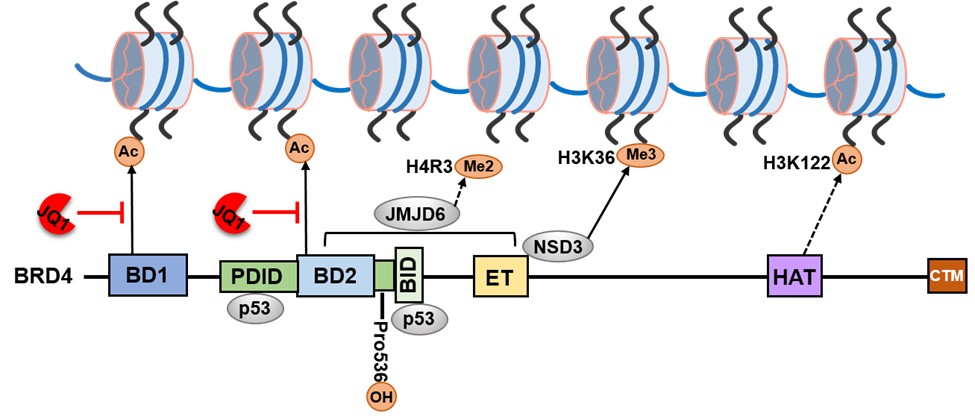
Figure 2. Interaction partners and modifications of BRD4. BRD4 contains several domains that recruit and interact with different proteins. BD1 and BD2 domains bind acetylated lysine of histone tails. The “pan” BET inhibitor JQ1 can engage at both BD1 and BD2 domains competitively with acetylated lysine binding. The PDID and BID domains directly bind p53. The ET domain recruits arginine demethylase JMJD6, which demethylates H4R3, and histone methyltransferase NSD3, which trimethylates H3K36. BRD4 has been characterized as a histone acetyltransferase with a putative HAT domain that acetylates H3K122. Prolyl hydroxylase domain protein 2 (PHD2) mediates proline hydroxylation of BRD4 at Pro536. Ac: Acetylated lysine; BD: Bromodomain. BID: Basic residue–enriched Interaction Domain; ET: Extra-Terminal domain; Me2: Dimethylated arginine; Me3: Trimethylated lysine; PDID: Phosphorylation-Dependent Interaction Domain; HAT: Histone Acetyltransferase; H4R3: Histone 4 Arginine 3; H3K36: Histone 3 Lysine 36; H3K122:Histone 3 Lysine 122; JMJD6: Jumonji C-domain-containing protein 6; NSD3: Histone-lysine N-methyltransferase.
Translational Potential for BET Inhibitors in Human Heart Disease
Based on the promising effects seen in the preclinical studies, prospective cardiovascular clinical trials (phase II and III) investigating the BET inhibitor RVX-208 (now called apabetalone) have already been completed. The phase II ASSERT trial (ApoA-I Synthesis Stimulation Evaluation in Patients Requiring Treatment for Coronary Artery Disease) represented the first attempt to assess the safety, tolerability, and efficacy of RVX-208 in patients with stable coronary artery disease who were on statin therapy [37]. Compared to placebo, administration of RVX-208 significantly increased levels of high-density lipoprotein cholesterol (HDL-C) and large HDL particles, suggesting improved cholesterol mobilization. However, there was no statistical significance regarding apolipoprotein A1 (ApoA1) changes, and transient and reversible elevations in liver transaminases were observed. The ASSERT trial was limited by a short treatment window of only 12 weeks. Later, a 26-week phase II ASSURE trial (ApoA-I Synthesis Stimulation and Intravascular Ultrasound for Coronary Atheroma Regression Evaluation) showed that RVX-208 failed to significantly elevate ApoA1 and HDL-C and failed to significantly decrease the primary endpoint of percent change in atheroma volume [38]. The beneficial effect of RVX-208 on facilitation of cholesterol mobilization was lost in the ASSURE trial, which may indicate the importance of combination drug therapy. The recently completed phase III BETonMACE trial (major adverse cardiovascular events) is a randomized, doubleblind, placebo-controlled trial conducted at 190 sites in 13 countries. Overall, 2425 patients with acute coronary syndrome, T2DM, and low HDL-C levels, participated in the trial [39]. Although not reaching significance on the primary endpoint (combined cardiovascular death, MI, or stroke), apabetalone treatment showed a trend toward reduction in MACE (10.3% in apabetalone-treated and 12.4% in placebo-treated patients, hazard ratio 0.82, 95% confidence interval [CI] 0.65–1.04), among the 2425 subjects. The authors suggested that a larger trial with expanded endpoints may yield a different result. Notably, apabetalone is a BET inhibitor selective for the second BD (BD2) within the BET proteins. This selective inhibition of BD2 by apabetalone largely contributes to its well described safety and tolerability profile, which far exceeds those of pan-BET inhibitors [40]. Overall, the sample size, the sample selection criteria, the treatment window, and the combination drug therapy will all influence the results of the clinical trials. In addition, given the diverse effects of BET proteins on mammalian physiology and pathophysiology, it will be possible that the side effects induced by BET inhibitor will also influence the outcome of clinical trials. Although the preclinical studies showed promising effect of BET inhibition, the current clinical advancements of BET inhibitors in cardiovascular diseases are still in early stages.
Perspectives
Recent preclinical studies have identified the indispensable role of BRD4 in mouse heart and highlighted the therapeutic benefits of BET BD inhibition in various types of heart disease, including MI or TAC-induced HF, gene mutation-induced DCM, and DC. HF in patients is often categorized as reduced ejection fraction (HFrEF) or preserved ejection fraction (HFpEF). Patients with HFpEF also have poor prognosis after the first diagnosis [41]. Whereas multiple studies have focused on investigating the role of BET BD inhibition in HFrEF, exploring its function in HFpEF will be of significance. Moreover, acute HF is often associated with ischemia-reperfusion injury [42]. Investigating the role of BET inhibition during ischemia-reperfusion is also of interest. Clinical trials of the BD2-selective BET inhibitor apabetalone in cardiovascular diseases suggest significant potential for future development. Despite these exciting advances, many questions remain to be answered. What are the distinct roles of individual BET proteins in the setting of different cell types in the heart? Is it possible to study the BET-independent function of BET proteins in different types of heart disease? How can more potent BD-selective compounds be developed? In conclusion, an in-depth understanding of the precise mechanisms underlying BET BD inhibition and discovery of more potent inhibitory compounds will foster the development of BET inhibitors as therapeutic strategies for human heart disease.
References
2. Jiang YW, Veschambre P, Erdjument-Bromage H, Tempst P, Conaway JW, Conaway RC, et al. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proceedings of the National Academy of Sciences. 1998 Jul 21;95(15):8538-43.
3. Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, et al. Selective inhibition of BET bromodomains. Nature. 2010 Dec;468(7327):1067-73.
4. Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010 Dec;468(7327):1119-23.
5. Das S, Senapati P, Chen Z, Reddy MA, Ganguly R, Lanting L, et al. Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells. Nature Communications. 2017 Nov 13;8(1):1-9.
6. Meloche J, Potus F, Vaillancourt M, Bourgeois A, Johnson I, Deschamps L, et al. Bromodomaincontaining protein 4: the epigenetic origin of pulmonary arterial hypertension. Circulation Research. 2015 Aug 28;117(6):525-35.
7. Brown JD, Lin CY, Duan Q, Griffin G, Federation AJ, Paranal RM, et al. NF-κB directs dynamic super enhancer formation in inflammation and atherogenesis. Molecular Cell. 2014 Oct 23;56(2):219-31.
8. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart disease and stroke statistics—2019 update: a report from the American Heart Association. Circulation. 2019 Mar 5;139(10):e56-28.
9. Hill JA, Olson EN. Cardiac plasticity. New England Journal of Medicine. 2008 Mar 27;358(13):1370-80.
10. Spiltoir JI, Stratton MS, Cavasin MA, Demos-Davies K, Reid BG, Qi J, et al. BET acetyl-lysine binding proteins control pathological cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2013 Oct 1;63:175-9.
11. Anand P, Brown JD, Lin CY, Qi J, Zhang R, Artero PC, et al. BET bromodomains mediate transcriptional pause release in heart failure. Cell. 2013 Aug 1;154(3):569-82.
12. Stratton MS, Lin CY, Anand P, Tatman PD, Ferguson BS, Wickers ST, et al. Signal-dependent recruitment of BRD4 to cardiomyocyte super-enhancers is suppressed by a microRNA. Cell Reports. 2016 Aug 2;16(5):1366-78.
13. Duan Q, McMahon S, Anand P, Shah H, Thomas S, Salunga HT, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Science Translational Medicine. 2017 May 17;9(390).
14. Stratton MS, Bagchi RA, Felisbino MB, Hirsch RA, Smith HE, Riching AS, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circulation Research. 2019 Sep 13;125(7):662-77.
15. Stratton MS, Haldar SM, McKinsey TA. BRD4 inhibition for the treatment of pathological organ fibrosis. F1000Research. 2017;6.
16. Mills RJ, Humphrey SJ, Fortuna PR, Lor M, Foster SR, Quaife-Ryan GA, et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARSCoV- 2 infection. Cell. 2021 Apr 15;184(8):2167-82.
17. Schultheiss HP, Fairweather D, Caforio AL, Escher F, Hershberger RE, Lipshultz SE, et al. Dilated cardiomyopathy. Nature Reviews Disease Primers. 2019 May 9;5(1):1-9.
18. Stewart CL, Kozlov S, Fong LG, Young SG. Mouse models of the laminopathies. Experimental Cell Research. 2007 Jun 10;313(10):2144-56.
19. Auguste G, Rouhi L, Matkovich SJ, Coarfa C, Robertson MJ, Czernuszewicz G, et al. BET bromodomain inhibition attenuates cardiac phenotype in myocyte-specific lamin A/C–deficient mice. The Journal of Clinical Investigation. 2020 Sep 1;130(9):4740-58.
20. Schmitt JP, Kamisago M, Asahi M, Li GH, Ahmad F, Mende U, et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science. 2003 Feb 28;299(5611):1410-3.
21. Antolic A, Wakimoto H, Jiao Z, Gorham JM, DePalma SR, Lemieux ME, et al. BET bromodomain proteins regulate transcriptional reprogramming in genetic dilated cardiomyopathy. JCI Insight. 2020 Aug 6;5(15).
22. Tan Y, Zhang Z, Zheng C, Wintergerst KA, Keller BB, Cai L. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nature Reviews Cardiology. 2020 Sep;17(9):585-607.
23. Guo M, Wang HX, Chen WJ. BET-inhibition by JQ1 alleviates streptozotocin-induced diabetic cardiomyopathy. Toxicology and Applied Pharmacology. 2018 Aug 1;352:9-18.
24. Mu J, Zhang D, Tian Y, Xie Z, Zou MH. BRD4 inhibition by JQ1 prevents high-fat diet-induced diabetic cardiomyopathy by activating PINK1/Parkin-mediated mitophagy in vivo. Journal of Molecular and Cellular Cardiology. 2020 Dec 1;149:1-4.
25. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. New England Journal of Medicine. 2007 Jun 14;356(24):2457-71.
26. Barth E, Stämmler G, Speiser B, Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. Journal of Molecular and Cellular Cardiology. 1992 Jul 1;24(7):669-81.
27. Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, et al. Mitochondrial function as a therapeutic target in heart failure. Nature Reviews Cardiology. 2017 Apr;14(4):238-50.
28. Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010 Feb;12(2):119-31.
29. Padmanabhan A, Alexanian M, Linares-Saldana R, González-Terán B, Andreoletti G, Huang Y, et al. BRD4 (bromodomain-containing protein 4) interacts with GATA4 (GATA binding protein 4) to govern mitochondrial homeostasis in adult cardiomyocytes. Circulation. 2020 Dec 15;142(24):2338-55.
30. Kim SY, Zhang X, Schiattarella GG, Altamirano F, Ramos TA, French KM, et al. Epigenetic reader BRD4 (bromodomain-containing protein 4) governs nucleusencoded mitochondrial transcriptome to regulate cardiac function. Circulation. 2020 Dec 15;142(24):2356-70.
31. Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 2013 Dec 19;155(7):1581-95.
32. Rahman S, Sowa ME, Ottinger M, Smith JA, Shi Y, Harper JW, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Molecular and Cellular Biology. 2011 Jul 1;31(13):2641-52.
33. Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers Brd4 chromatin binding and activator recruitment for gene-specific targeting. Molecular Cell. 2013 Mar 7;49(5):843-57.
34. Devaiah BN, Case-Borden C, Gegonne A, Hsu CH, Chen Q, Meerzaman D, et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nature Structural & Molecular Biology. 2016 Jun;23(6):540-8.
35. Erber L, Luo A, Chen Y. Targeted and Interactome Proteomics Revealed the Role of PHD2 in Regulating BRD4 Proline Hydroxylation*[S]. Molecular & Cellular Proteomics. 2019 Sep 1;18(9):1772-81.
36. Zhou T, Erber L, Liu B, Gao Y, Ruan HB, Chen Y. Proteomic analysis reveals diverse proline hydroxylationmediated oxygen-sensing cellular pathways in cancer cells. Oncotarget. 2016 Nov 29;7(48):79154.
37. Nicholls SJ, Gordon A, Johansson J, Wolski K, Ballantyne CM, Kastelein JJ, et al. Efficacy and safety of a novel oral inducer of apolipoprotein AI synthesis in statin-treated patients with stable coronary artery disease: a randomized controlled trial. Journal of the American College of Cardiology. 2011 Mar 1;57(9):1111-9.
38. Nicholls SJ, Puri R, Wolski K, Ballantyne CM, Barter PJ, Brewer HB, et al. Effect of the BET protein inhibitor, RVX-208, on progression of coronary atherosclerosis: results of the phase 2b, randomized, double-blind, multicenter, ASSURE trial. American Journal of Cardiovascular Drugs.2016 Feb 1;16(1):55-65.
39. Ray KK, Nicholls SJ, Buhr KA, Ginsberg HN, Johansson JO, Kalantar-Zadeh K, et al. Effect of apabetalone added to standard therapy on major adverse cardiovascular events in patients with recent acute coronary syndrome and type 2 diabetes: a randomized clinical trial. JAMA. 2020 Apr 28;323(16):1565-73.
40. Kulikowski E, Rakai BD, Wong NC. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Medicinal Research Reviews. 2021 Jan;41(1):223-45.
41. Meta-analysis Global Group in Chronic Heart Failure (MAGGIC). The survival of patients with heart failure with preserved or reduced left ventricular ejection fraction: an individual patient data meta-analysis. European Heart Journal. 2012 Jul 1;33(14):1750-7.
42. Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cellular Physiology and Biochemistry. 2018;46(4):1650-67.